《有机合成-切断法》章末总结1
有机基础知识学了一遍之后来看点合成的内容,首先是这本比较基础的《切断法》。
在正式开始之前先随便聊几点。
一是《有机合成——切断法》这本书的选择,我看的是中译本(ISBN: 9787030276704),由药明康德公司翻译出版,但是恕我直言,翻译水平是真烂。我相信绝不是药明康德公司的研发人员没水平,纯粹是敷衍了事随便写写而已,书中有大量的句子表述极为生硬,完全就是没有好好思考、完全按英语语序直译的结果,我相信就算是拿AI润色一下也会比现在好得多。
这都不算严重的问题了,毕竟文本只要能把意思传达到位就可以,但是该中译本的印刷质量更是灾难,书中的错别字/印刷错误比比皆是,有些严重影响到了文意理解,例如字母I和T还有数字1经常混淆,会出现如$CHC1_3$这种尴尬的东西,更离谱的是把有一处参考文献中作者的姓从T印刷为I,导致文献找不到。
除了翻译问题,原书中也是有不少事实错误,例如中译本205页参考文献19处对于反应现象的描述与原文献中恰好相反,再如中译本271页参考文献10处所画的反应式中,反应物比生成物少了个碳。之前我听过许多人都不太推荐这本书,在我自己看完之后,我仍然觉得这是一本很好的有机合成入门教材,但是其中各种大大小小的错误实在是败坏看书的心情。
二是笔记整理的方式,对于这本书来说,内容大多仍是比较基础的,所以不会每一章都写,只会挑部分重要内容整理和记录,并且附上几个我决定不错的例子,所以这一个series的数字并不对应原书的章节数(毕竟这本书有40章),只是一个顺序的表示而已。在这一series中,我也会尝试输出更多的个人的总结和思考,不再只是一味抄书记录了。
所以后面就随自己的感觉写了,与原书的章节没有强相关,但是先后顺序是一样的。
1 基本术语
如标题所言,切断是有机合成的必备前置步骤,对于一个复杂的目标分子,合理的、优秀的切断会让合成的思路以及具体实施变得更加轻松。为了方便表述,在最开始有必要介绍并约定本书使用的基本术语和表示方法。
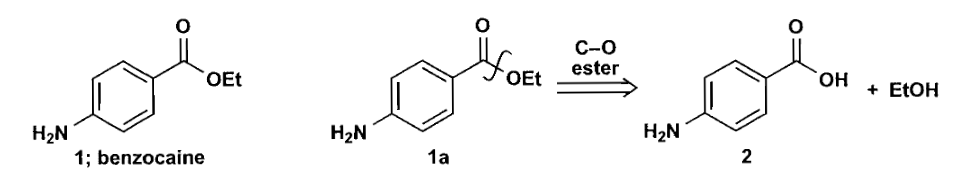
在对目标分子的分析时,你可以用波浪线来表示对某一根化学键的切断,用双线箭头来表示切断的进行,即得到了某一步反应的反应物,箭头上可以写出切断化学键的种类以及反应类型。看一个例子:
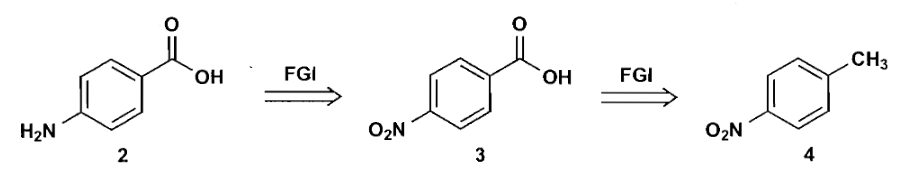
在逆合成分析中,你有时需要对官能团进行转化以进行后续的分析,这一步被称为官能团转换(functional group interconversion, FGI)。
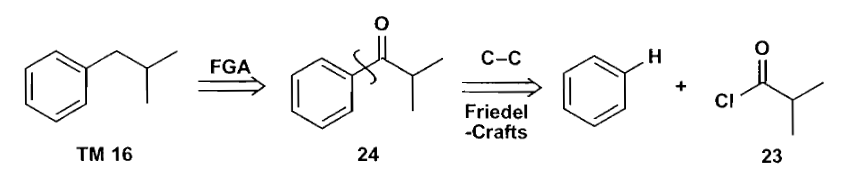
有时我们则需要人为给分子加上一些官能团/结构,这样才利于反应的发生或调控选择性等,这一步被称为官能团添加(functional group addition, FGA)。
在切断时,如果把化合物切断为两个部分,那么这两个带有电荷的片段就被称作合成子(synthons)。
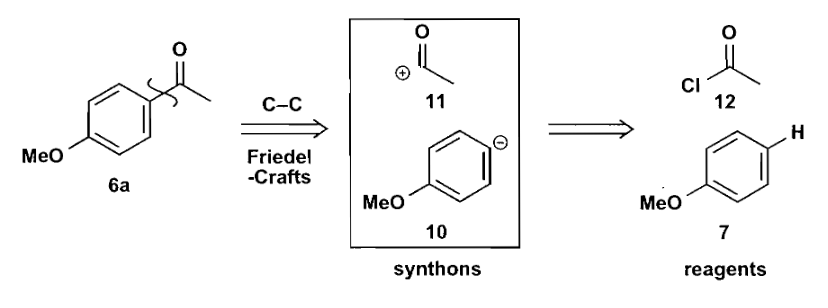
合成子可以是反应过程中确确实实存在的中间体,也可以只是一种不存在的表示,合成子可以给你提供一些试剂选用的思路,当然在熟练之后也可以不画出合成子。
2 单官能团切断
最简单的切断法就是单官能团的C-X键切断了,相对应的反应主要是$S_N1, S_N2$亲核取代,以及羧酸衍生物的转化反应等。合成子的选用都是比较常见的物质。
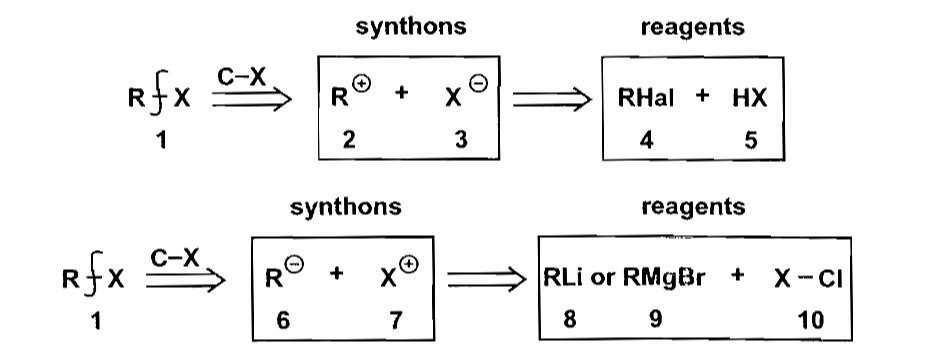
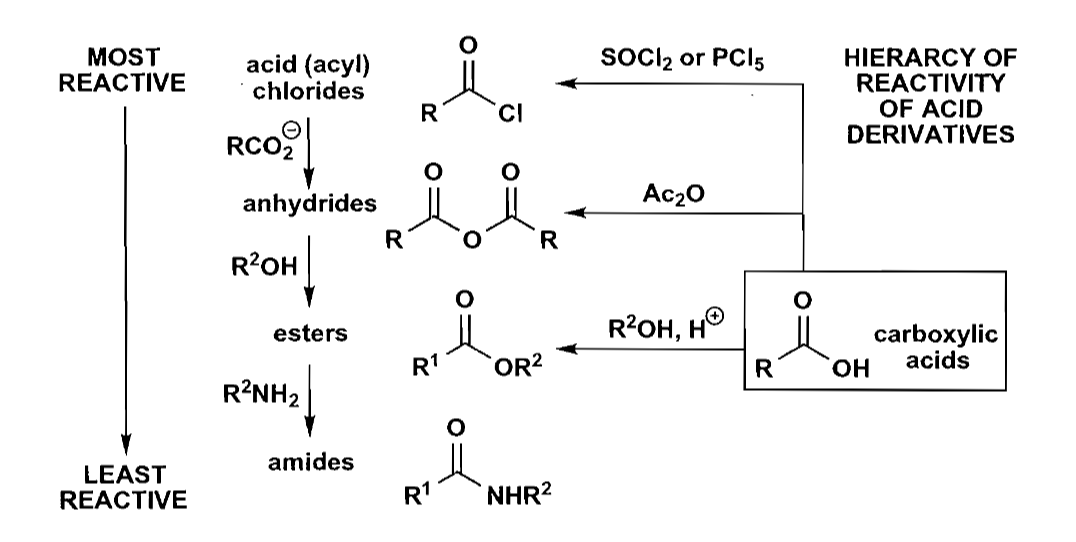
羧酸衍生物的互相转化也是比较基础的内容,没什么好多说的。
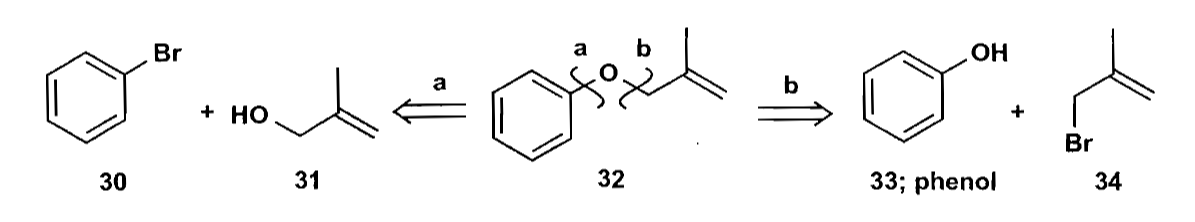
切断时需要考虑切断的具体路径,不同的切断方式有优劣之分,甚至一些路径并不可行,如下例。切断途径a涉及芳香亲核取代,实际上无法进行。
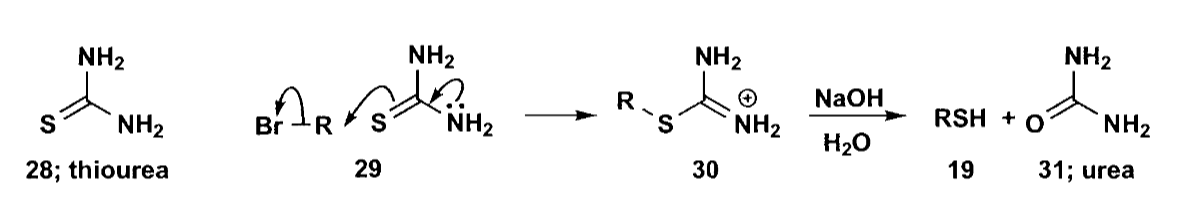
在使用亲核取代反应合成目标化合物时,需要考虑竞争反应的问题,对于竞争反应可以通过保护基来选择性的反应。例如,使用硫化物和卤代烷烃反应制备硫醇的反应很难控制,容易过度反应生成硫醚。但是如果我们用硫脲替代$NaSH$作为亲核试剂,那么竞争反应就可以不必考虑,可以很好的制得硫醇。
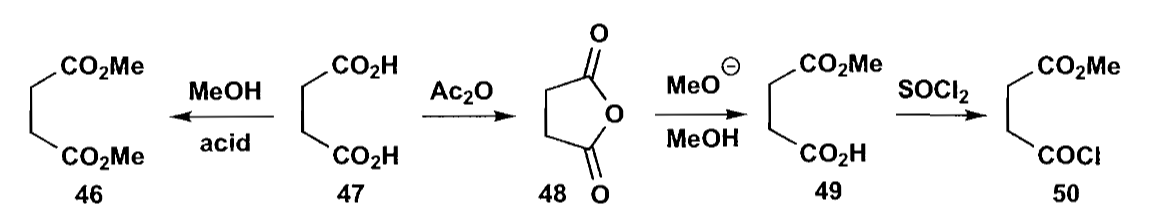
如果同时存在两个等同的官能团需要选择性反应时,另一个策略是把两个基团合成一个。一个典型的例子就是内酸酐的单官能团转化。
3 二官能团切断
如果你要合成如下的硫醚化合物,一个方法是硫醇的亲核反应。
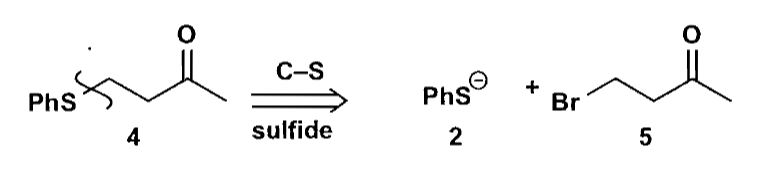
这个方法有一个缺点:没有考虑到硫醇对羰基的竞争反应。更好的一个策略是,使用二官能团切断,利用硫醇和不饱和酮的共轭加成完成反应。
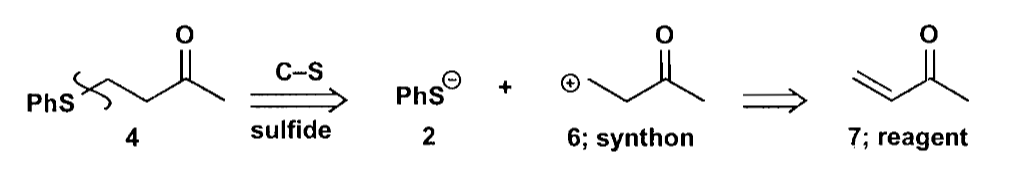
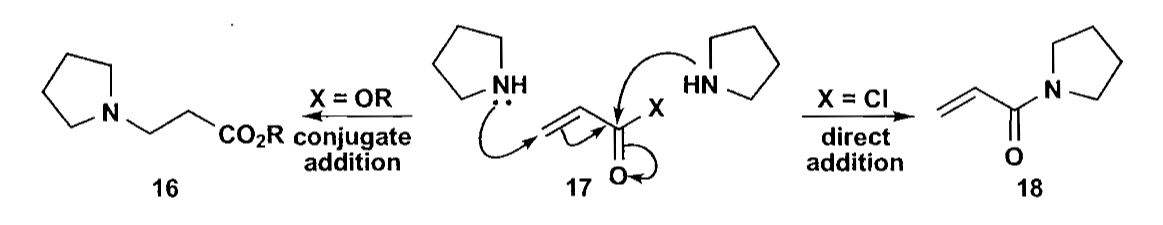
如上的1,3-二官能团切断法就是非常常见的利用亲核试剂+不饱和酮的共轭加成例子,这种方法需要考虑加成的区域选择性,确保我们能够得到1,4-加成产物。一般来说,亲电性强的化合物(酰氯、醛)会生成羰基加成产物;亲电性弱的化合物(酯、酮)倾向与共轭加成产物。
1,2-官能团切断可以切开为常规的杂原子亲核试剂和一个亲电合成子,对于醇类的合成子来说,一个非常好用的试剂是环氧化合物。
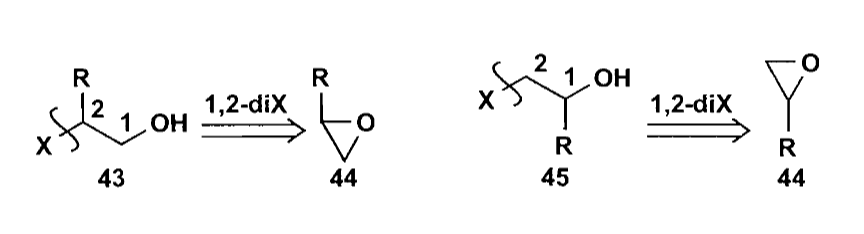
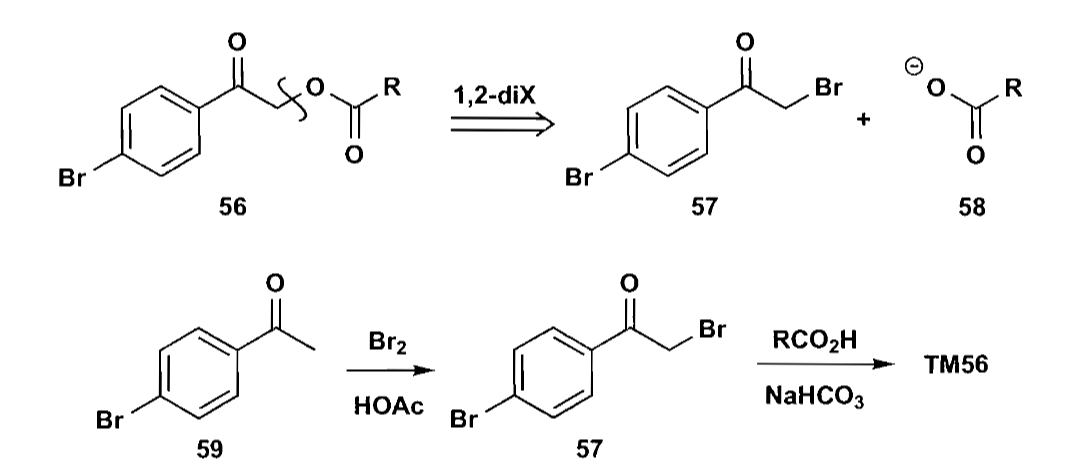
羰基化合物可以切开为烯醇化合物和亲电合成子,最常见的例子是羰基化合物的α-卤代。同时α-卤代羰基化合物也可以作为亲电试剂被进攻。
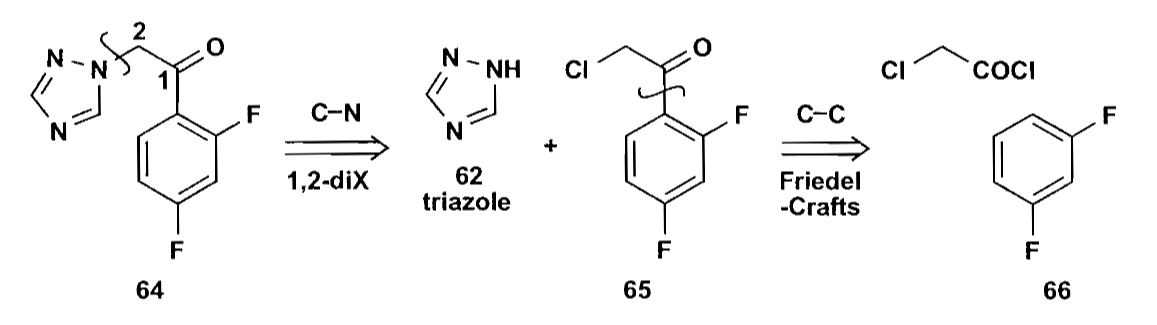
最后看个例子,药物氟康唑Fluconazole的合成的部分路线如下,仍然是类似的对α-卤代羰基化合物的亲核取代。
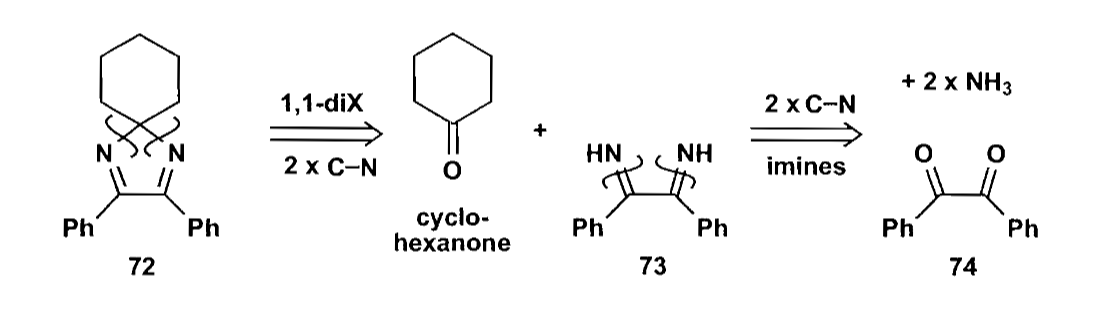
1,1-二官能团的切断常见与缩醛及其类似结构,在保护基的应用中可以说是最为基础的反应。
4 胺与醇
胺的合成不能使用亲核取代来直接制备,因为烷基取代的胺会具有更强的取代能力,会使反应进行到底。
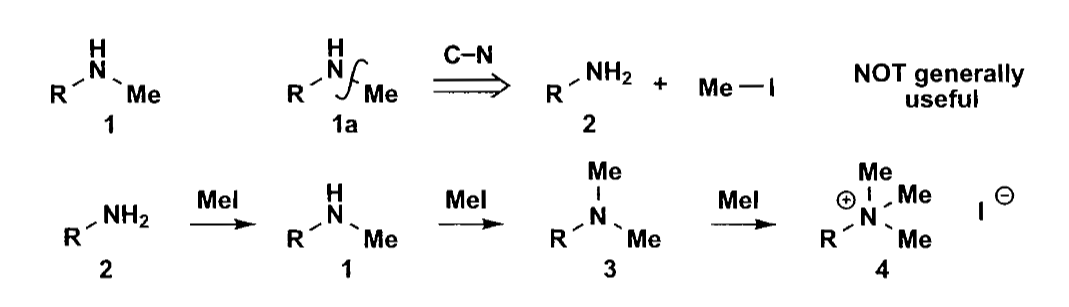
所以胺的制备需要采用其他方案,一个很好的选择是还原胺化反应,可以说是制备胺类化合物最重要的方法之一。亚胺的C=N双键比醛酮的C=O键弱,因此我们可以选用恰当的试剂做到选择性还原。在弱酸条件下,亚胺盐的亲电性更强,易于与还原剂反应,但是一般的硼氢化钠等还原剂在酸性条件下不稳定,所以还原胺化中常用的试剂主要是$[NaB(CN)H_3]$或$[NaB(OAc)_3H]$,吸电子基团可以稳定硼原子上的负氢的亲核性,使之能选择性的还原亚胺,并能在酸中稳定存在。
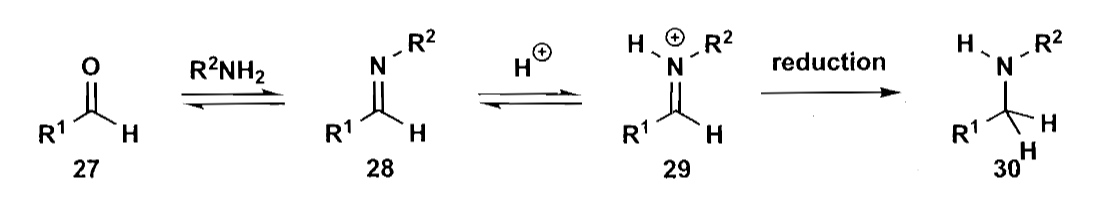
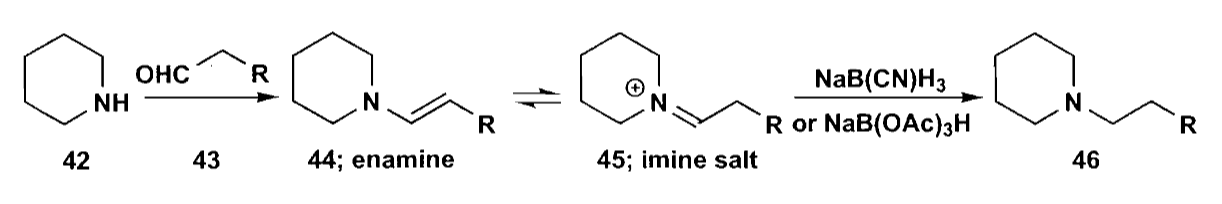
这里有一点需要注意,还原胺化也可以制备叔胺,虽然看起来叔胺只能形成烯胺,但是叔胺也可以形成同样的亚胺盐结构,而这正是选择性还原所需要的。
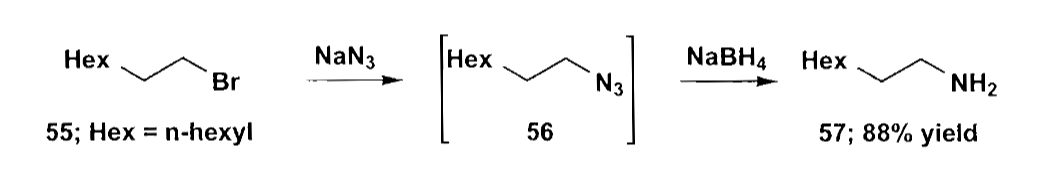
除了还原胺化以外,制备胺的方法还有含氮亲核试剂的烷基化,例如叠氮或氰基。
烷基化反应对于有大位阻基团的胺的合成比较吃力,这时我们可以考虑Ritter反应,可以制备与叔碳原子相连的胺。
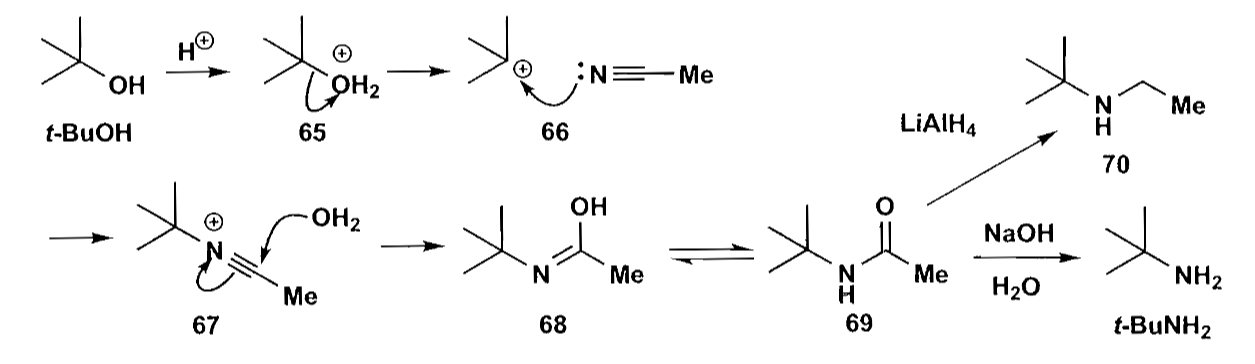
接下来让我们看看醇的合成,一类重要的策略是羰基化合物的转化,这将涉及到1,1 C-C键的切断。
醇可以通过氧化反应制备得到醛酮、羧酸及其衍生物,这部分内容较为基础,可以查阅基础有机,此处按下不表。
还有1,2 C-C键的切断也可以制备醇类,最常见的就是环氧化合物的开环。
5 烯烃
烯烃可以在酸性条件下由醇脱水制得,用于脱水的酸的酸性应当相当强,且没有亲核性离子。烷基卤代物的消除需要在碱条件下进行,同样的,为了避免亲核取代,应当使用大位阻的碱攫氢消除。
除了基础的消除反应,烯烃的合成的最重要方法是Wittig反应,通过选用合适的试剂可以较好地控制烯烃的构型。如果ylid的碳负离子可以被相连的吸电子基团稳定,则称该ylid为稳定ylid,反之则是不稳定ylid,这两种不同的ylid会生成不同构型的烯烃。
不稳定ylid会生成Z型烯烃,而稳定ylid会生成E型烯烃。一个很好的例子是白三烯抗体的合成,在一个流程中同时用到了E和Z的烯烃合成。
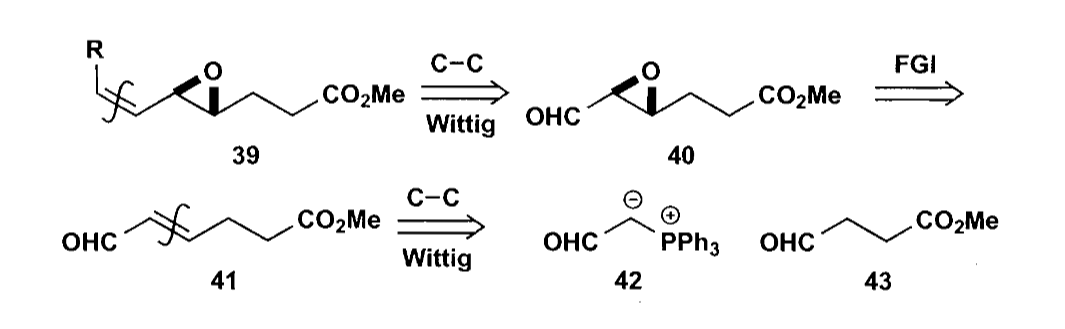
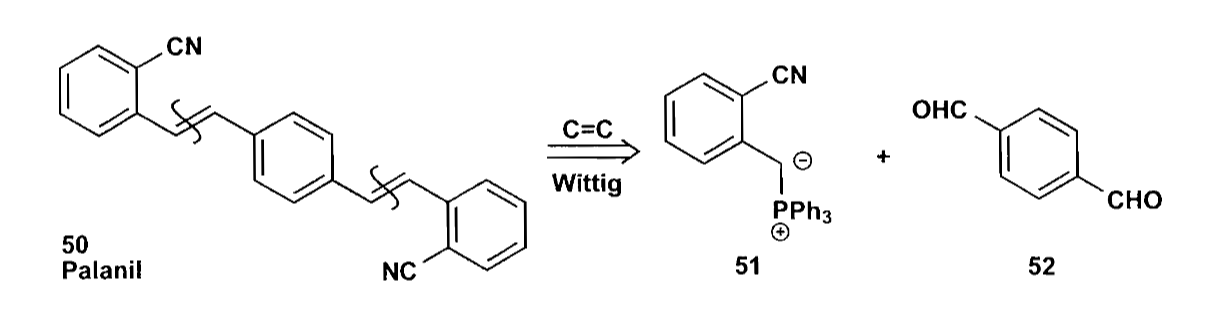
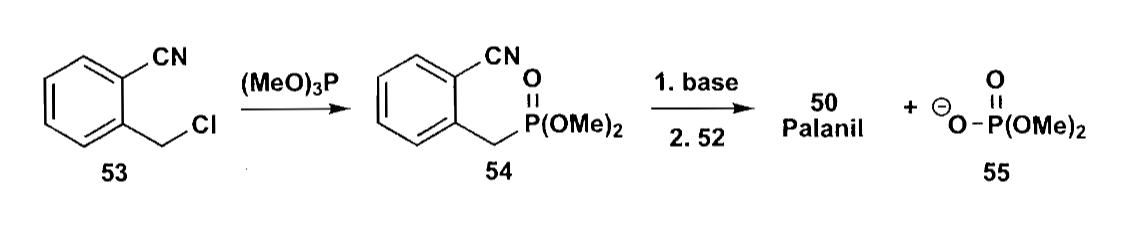
光学增白剂(Palanil)的合成也利用了稳定ylid的反应,使用易得的二醛原料合成了产物。但是ylid太过稳定也不太好,可能会无法与酮反应,因此一种改进方法是使用膦酸酯的阴离子进行Horner-Wadworth-Emmons(HWE)反应,该反应同样可以获得E型烯烃,可以看作是Wittig反应的改进。
总结
挑着写了一些,本书难度不大(尤其是前半部分),但是很多小细节还是值得一看的。