《中级有机化学》章末总结10
第十章介绍自由基和卡宾的反应,这些部分是基础有机里涉及较少的。
第10章 自由基及卡宾的反应
10.1 自由基引发方法
自由基反应为链式反应,过程分为:链引发(chain initiation)、链增长(chain propagation)和链终止(chain termination)。那么自由基反应要想发生,必须要通过链引发生成自由基。
常用的引发方式有四种,有高温裂解、自由基引发剂引发、光解均裂和单电子转移(single electron transfer, SET)。近年来发展的光催化反应还可以通过第一激发态三线态光催化剂分子的能量转移(energy transfer, ET)将底物分子转变为双自由基。下面主要介绍前四种常见方法。
高温裂解也称热解,常见于石油重整,在高温下发生C-C键的断裂,使重油变为轻油。均裂的难易程度取决于共价键的解离能(bond dissociation energy, BDE)。键解离能与键能有所不同,键解离能指绝对零度下键均裂所需能量,键能指分子中同一种共价键解离能的平均值。
举个例子,如甲烷中四个氢依次解离时,四根C-H键的键解离能不同,分别为435、444、444、339$KJ/mol$,C-H键键能即为四个值的平均值,为416$KJ/mol$。
然后是自由基引发剂。过氧化物中O-O单键在较低的温度下可以发生均裂,产生氧自由基,因此二叔丁基过氧化物、双氧水、过氧苯甲酰等物质可用作自由基引发剂。
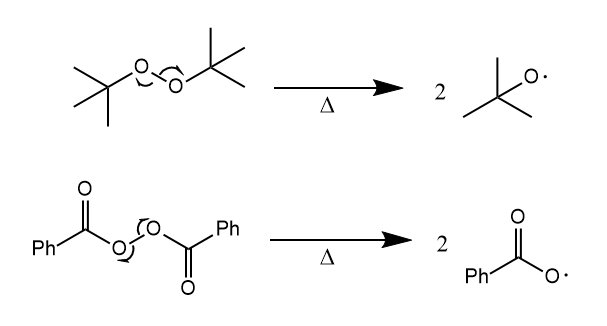
偶氮化合物对热敏感,加热条件下释放氮气生成烷基自由基。
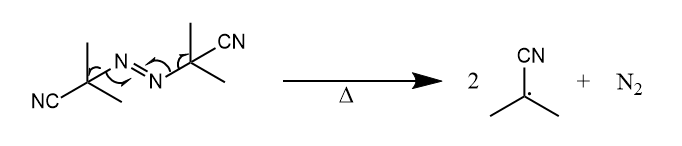
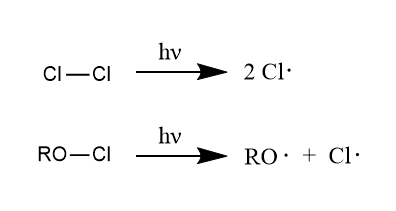
然后是光解均裂,光照条件下σ键的均裂是产生自由基的常用方法之一。
最后是单电子转移,一些无机金属离子具有氧化还原的性质,在氧化还原过程中势必涉及电子转移,如Fenton试剂。
$$
RO-OH +\ Fe^{2+} \rightarrow RO\cdot\ +\ OH^- +\ Fe^{3+}
$$
另一个例子是三烷基硼,它容易发生双分子均裂取代反应(bimolecular homolytic substitution, $S_H2$),这是产生自由基的常用方法之一。
$$
R_3B +\ O_2 \longrightarrow R\cdot +\ R_2BOO\cdot \[3mm]
R\cdot +\ O_2 \longrightarrow ROO\cdot \[3mm]
ROO\cdot +\ R_3B \longrightarrow R\cdot +\ R_2BOOR
$$
硼中心发生的$S_H2$反应是热力学有利的,因为B-O键的键解离能远大于B-C键的键解离能,二者之差约$175KJ/mol$。
10.2 自由基取代反应
最常见的一个反应就是基础有机中最先学到的烷烃的自由基卤化。包括链引发、链增长和链终止。
链引发:
$$
Cl-Cl \overset{h\nu}{\longrightarrow} \dot {Cl} +\ \dot {Cl}
$$
链转移:
$$
H_3C-CH_3 +\ \dot {Cl} \longrightarrow H_3C-CH_2 \cdot +\ HCl \[3mm]
H_3C-CH_2 \cdot +\ Cl-Cl \longrightarrow H_3C-CH_2Cl +\ \dot {Cl}
$$
链终止:
$$
H_3C-CH_2\cdot +\ \dot {Cl} \longrightarrow H_3C-CH_2Cl \[3mm]
H_3C-CH_2 \cdot +\ H_3C-CH_2 \cdot \longrightarrow H_3C-CH_2-CH_2-CH_3 \[3mm]
\dot{Cl} +\ \dot {Cl} \longrightarrow Cl_2
$$
烷烃中伯氢、仲氢和叔氢的反应性不同,伯氢最慢,叔氢最快,这与中间体自由基的稳定性相一致。
如果底物中含有双键或苯环,碳自由基能被双键和苯环稳定,如下所示反应是通过烯丙基自由基进行的。
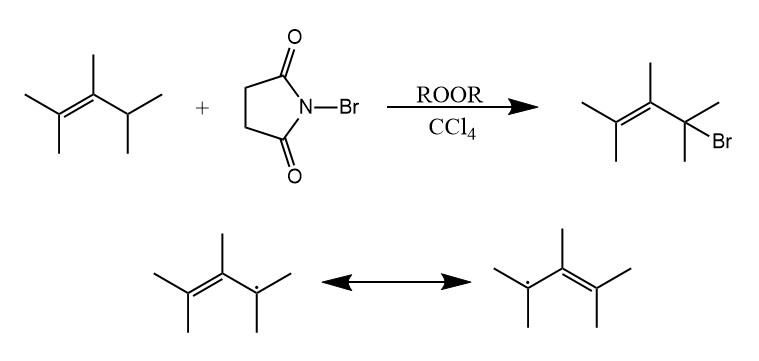
烷烃和卤素的相对反应速率大小是:氟>氯>溴>碘。其中氟代反应速率太快不易控制,碘代反应在能量上是不利的。氯和烷烃的反应速率大于溴和烷烃的反应速率,但是更快的反应速率意味着更低的选择性。
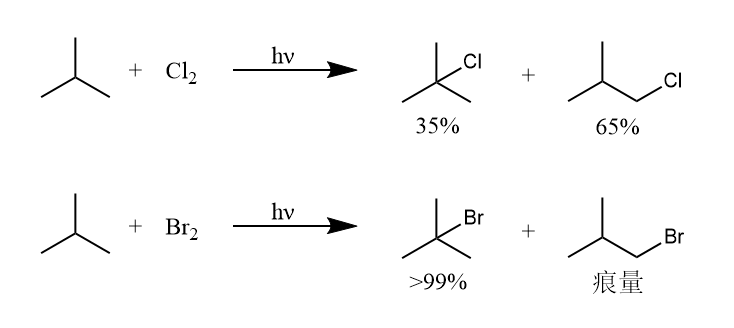
氯代反应是放能的,而溴代反应则是吸能的,因此根据Hommond假说,对于一个放能步骤,过渡态的结构更接近原料;对于一个吸能步骤,过渡态的结果更接近产物,由此可认为溴化反应经过的“类自由基过渡态”更接近产物的自由基,更具有自由基的性质。
换句话说,自由基的相对稳定性在溴化反应中更加重要,叔自由基的稳定性远高于伯自由基,所以自由基溴化反应的选择性表现很好。
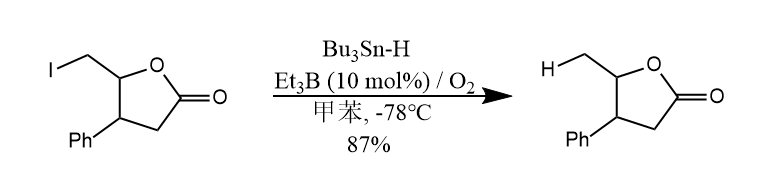
三丁基氢化锡和卤代烃反应,发生C-X键和Sn-H键的交换反应,生成C-X键还原产物,该反应在能量上是有利的。
$$
R-Br +\ Bu_3Sn-H \overset {h\nu}{\longrightarrow} R-H +\ Bu_3Sn-Br
$$
用催化量的三乙基硼和氧气作引发剂使得反应的条件更加缓和,极大地拓宽了底物的适用范围。
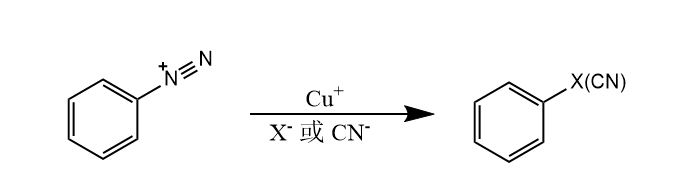
芳基重氮盐的C-N键均裂是产生芳基自由基的重要途径之一,例如Sandmeyer反应和Gomberg-Bachmann反应等。
铜(Ⅰ)盐催化下芳基重氮被负离子取代的反应称为Sandmeyer反应,负离子可以是$Cl^-, Br^-, CN^-, NO_2^-$等。
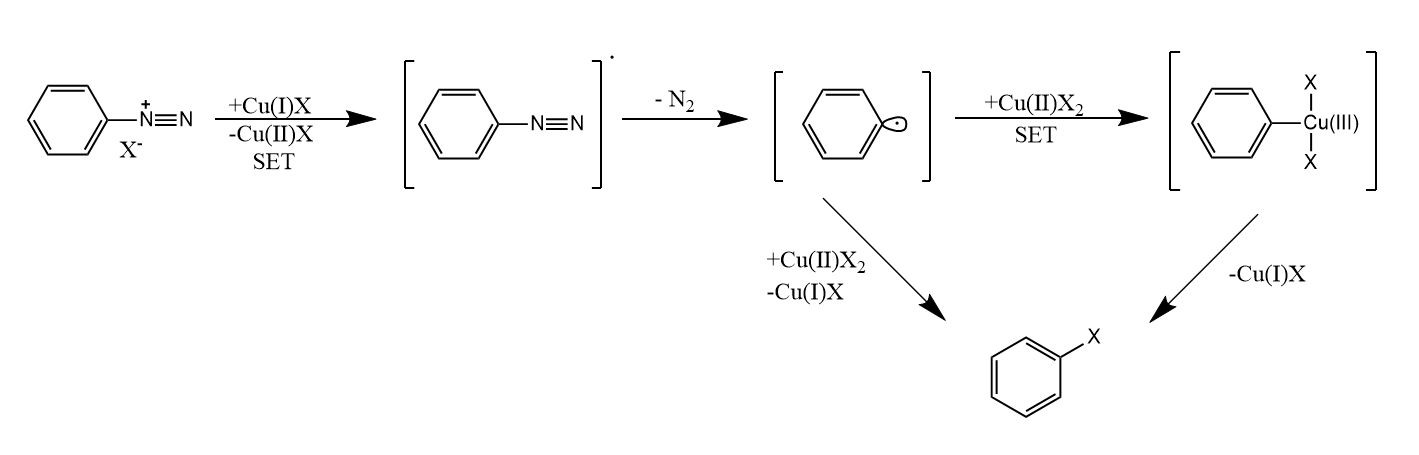
该反应通过芳基自由基机理进行,一价铜的SET过程向芳基重氮转移电子,生成芳基自由基和氮气,然后再次通过SET过程和铜(Ⅲ)配合物的还原消除得到产物。不过该反应是否经过三价铜中间体还有待证实。
利用Sandmeyer反应可以实现芳基重氮盐的三氟甲基化等有机化学中非常有用的转化。
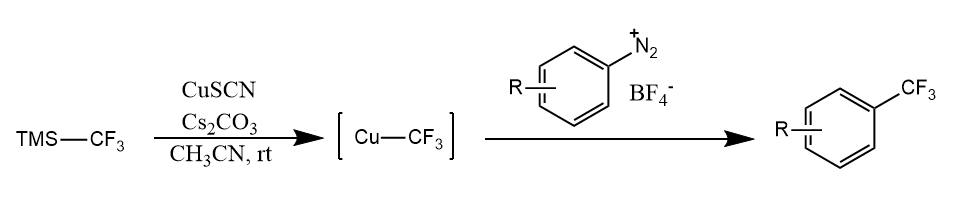
芳基重氮盐在碱性条件下能和芳烃偶联,生成联芳烃,这种反应称为Gomberg-Bachmann反应,此反应既可以在分子间发生,也可以在分子内发生。
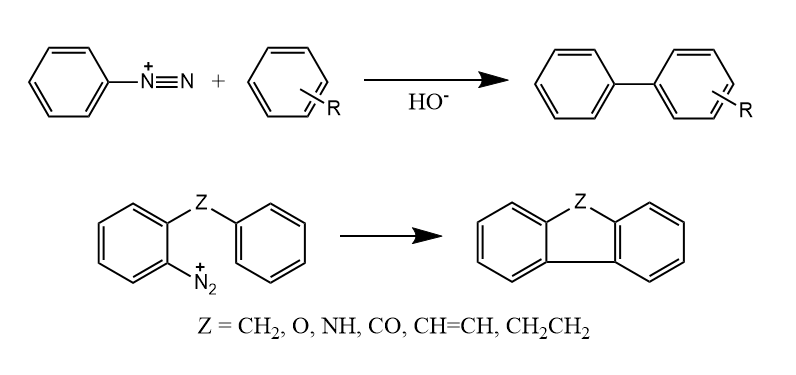
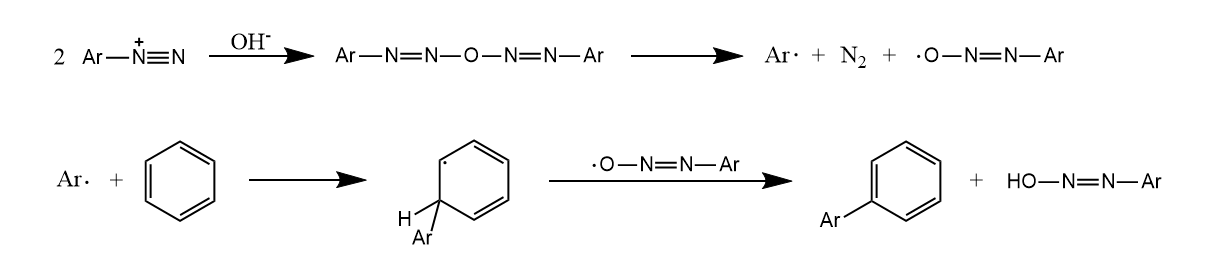
反应经历芳基自由基中间体,碱性条件的氢氧根参与反应生成的氧自由基推动了反应进行。
10.3 自由基加成反应
通常烯烃和溴化氢的加成生成马氏产物,但当混合物中有过氧化物存在时,反应生成反马氏产物,这是因为该反应是通过自由基机理进行的。
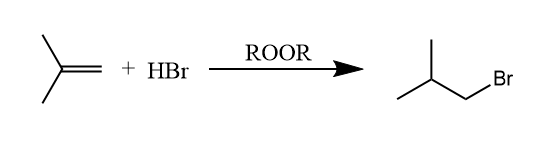
自由基中间体的稳定性决定了反应的区域选择性,因此得到反马氏产物也就不奇怪了。
烯烃和卤代烷的加成与加溴化氢类似,末端烯烃的加成具有区域选择性。
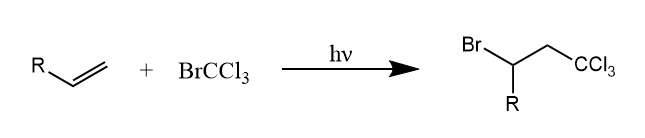
其机理第一步是卤代烷烃的均裂,生成两个自由基,如$Br\cdot$和$Cl_3C\cdot$,然后$Cl_3C\cdot$不可逆的加成到烯烃末端碳原子上形成C-C键,而$Br\cdot$的加成是可逆的,所以不会优先参与链增长。
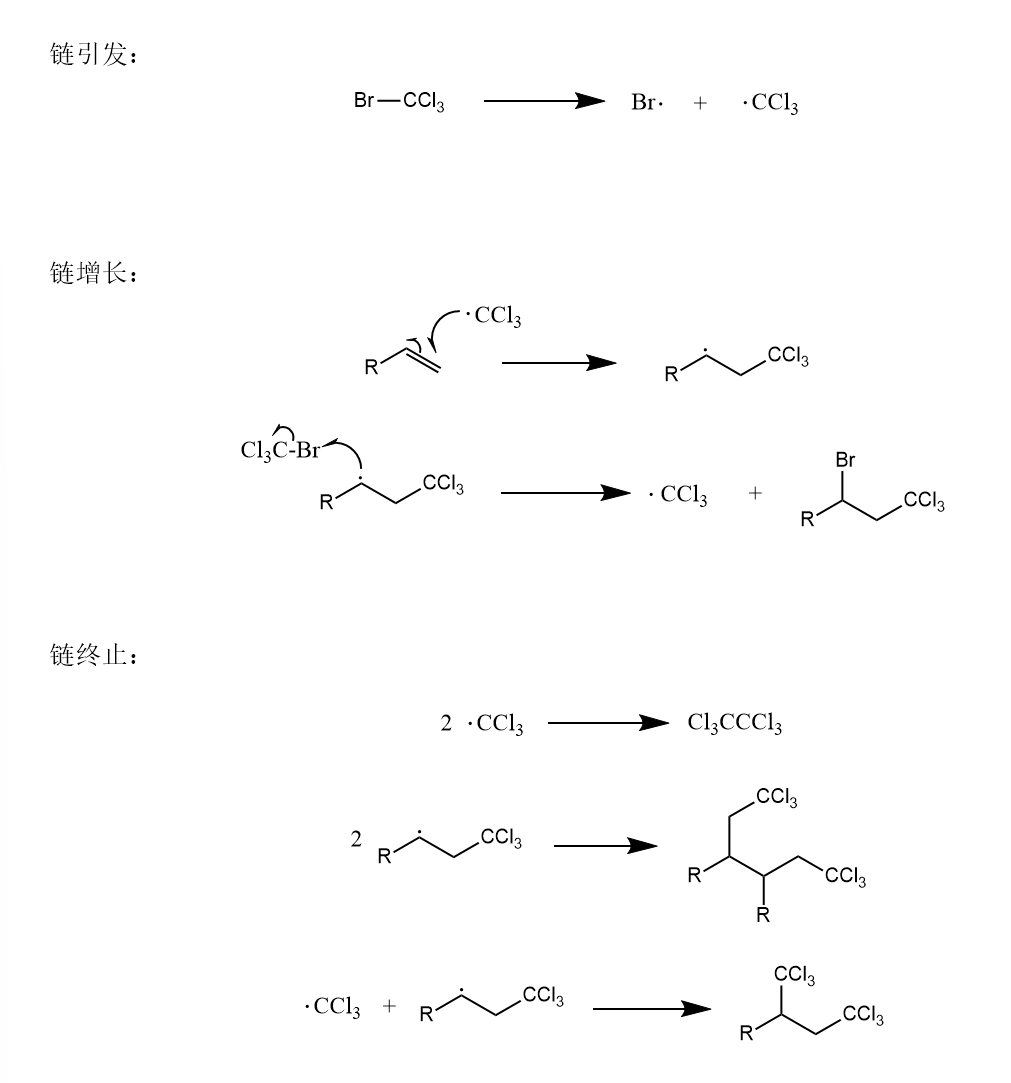
烯丙基溴化物的C-Br键容易均裂,在自由基引发剂作用下形成溴自由基对双键发生加成,该反应与上一个反应不同的是,溴自由基先参与链增长。
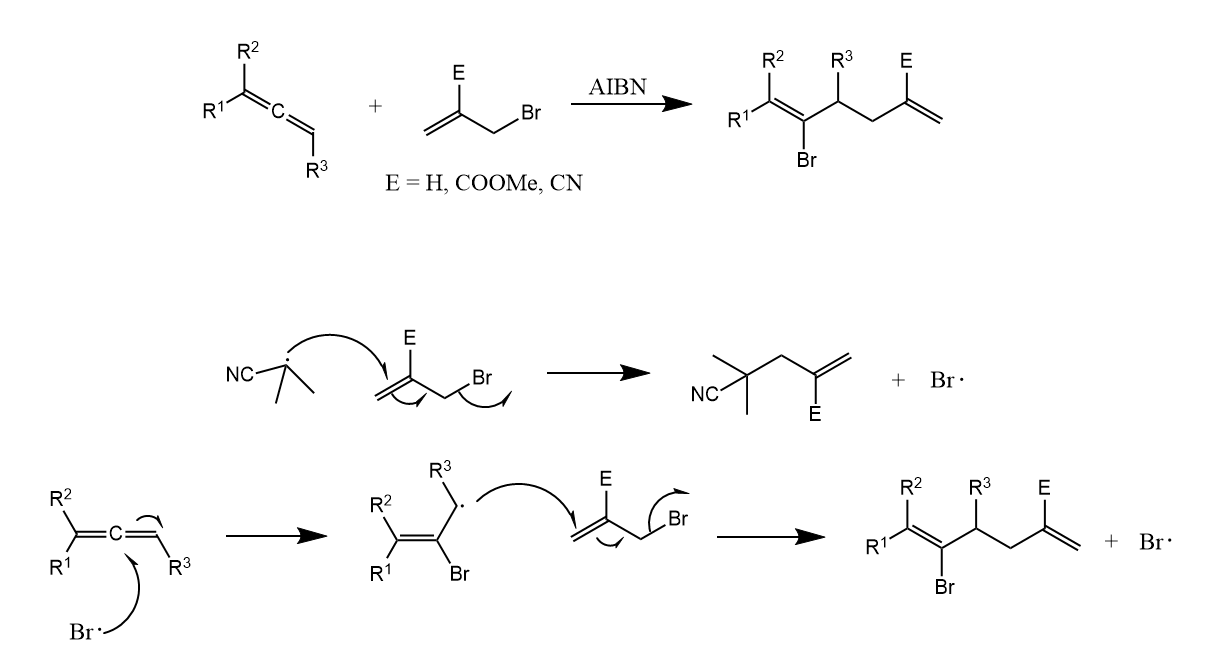
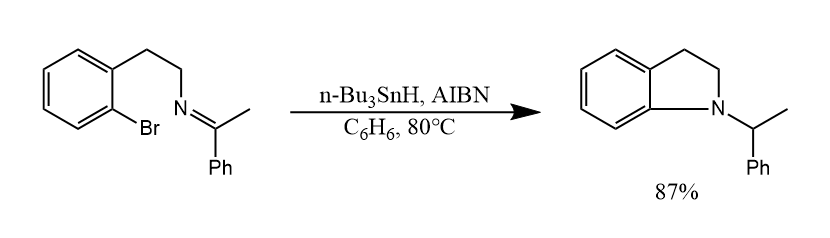
我们在10.2节中介绍了三丁基氢化锡对卤代烃的还原,如果在反应中加入烯烃,则可以将反应中的烷基自由基中间体捕获发生加成反应。
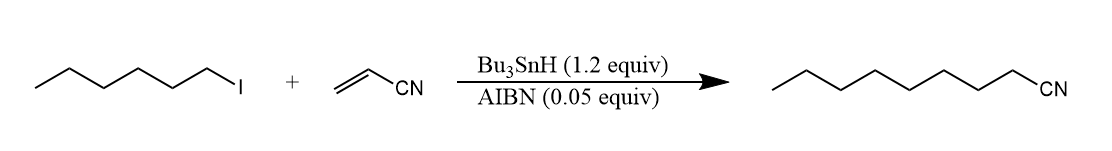
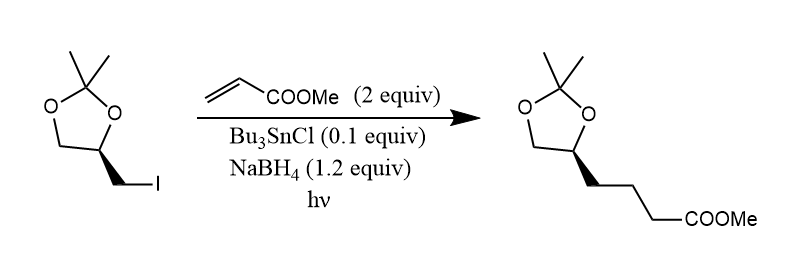
不难看出,上述反应中包含了还原和加成两种反应的竞争,而还原试剂(三丁基氢化锡)和加成试剂(烯烃)的比例决定了哪种反应占优,若要发生加成反应,则可增加烯烃的比例,或者降低三丁基氢化锡的比例。例如下面反应中,使用催化量的三丁基氢化锡,这样还原反应就会被抑制,同时加入过量硼氢化钠用于还原剂的再生。
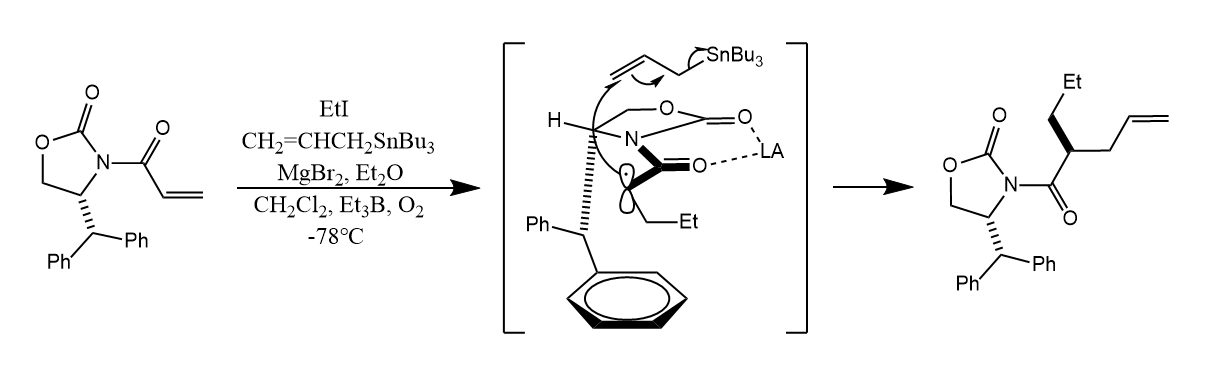
自由基加成的立体化学可以通过空间效应得到有效控制,使反应具有非对映选择性。例如下面的反应中,乙基自由基优先加成到双键上,烯丙基和新生成的自由基结合受到Lewis酸空间效应的约束,从远离苯基的一面加成。
分子内的自由基加成要比分子间的自由基反应容易的多,自由基所占$sp^3$轨道与$\pi^*$的有效重叠取决于链的长短。对于短链,自由基所占的$sp^3$轨道更容易和链相连的双键作用,因而优先得环较小的环化产物。这种区域选择性是由轨道方向性决定的,而与取代基种类和位置关系不大。
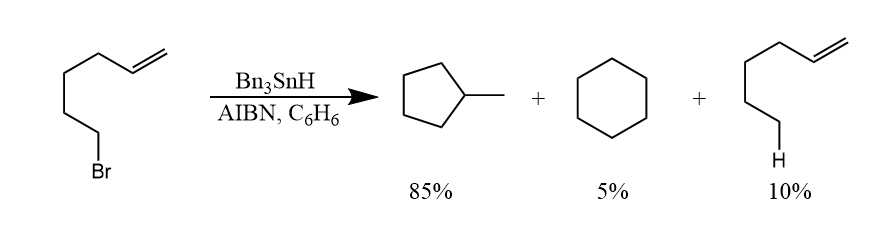
如上反应中,五元环产物为主要产物,而六元环产物仅占5%。
对于长链,反应的区域选择性还是取决于自由基中间体的性质,那么就与取代基的种类和位置有关了。
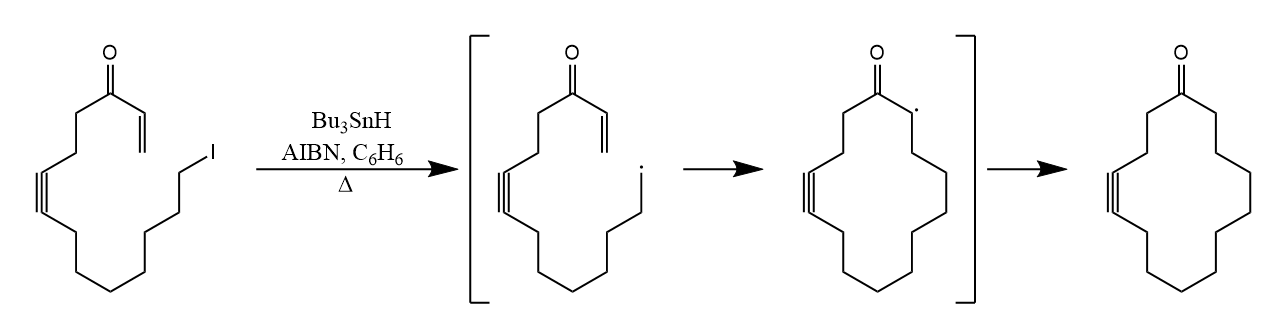
由于自由基加成的这种性质,其在五元杂环构建中起着重要作用,例如下面这个反应构建四氢吡咯衍生物,在反应历程中经历了分子内的自由基加成,并形成了较小的五元环。
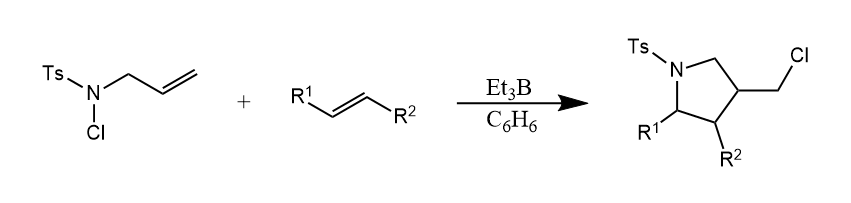
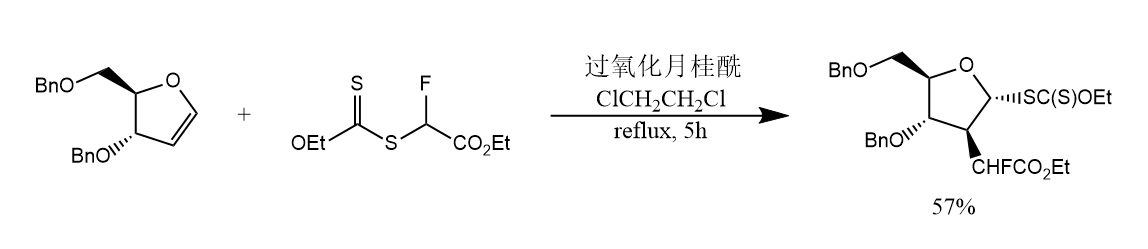
含硫化合物中的C-S键、S-S键和S-H键容易均裂,从而发生自由基反应。
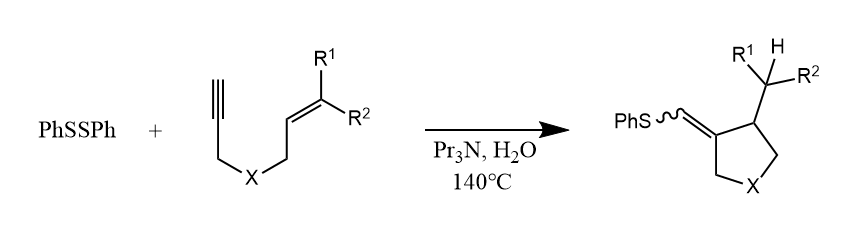
自由基也可对炔烃的碳碳三键发生加成,如:
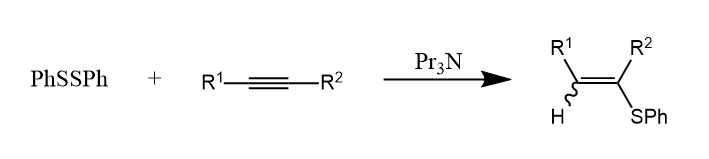
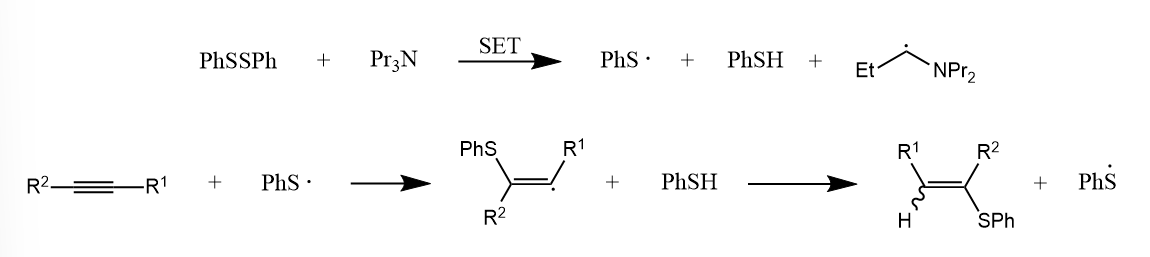
该反应通过SET过程产生苯硫自由基,继而发生自由加成。
如果底物中有多个不饱和键,生成的烯基自由基将分子内进攻空间是有利的不饱和键得到环合产物。
烃基自由基对于C=N双键的加成有两种区域选择性,也就是分别加成在C上或N上,通常情况下,C=N双键的自由基加成主要形成C-C键。如下例。
当然也有加成在N上的例子。
最后是自由基加成聚合反应,在高分子化学中占有及其重要的地位,60%以上的高分子材料是通过自由基加成聚合得到的。
自由基加成聚合包括链引发、链增长、链终止和链转移四个基元反应。自由基加成聚合反应的链增长过程有三个特征:一是放热、二是增长活化能低、三是聚合过程有选择性。链终止则有两种方式,分为耦合终止和歧化终止。
10.4 卡宾的形成及其反应
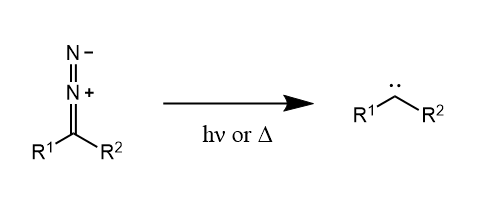
重氮化合物在光照或加热条件下可产生卡宾,这是卡宾中间体形成的主要方法。
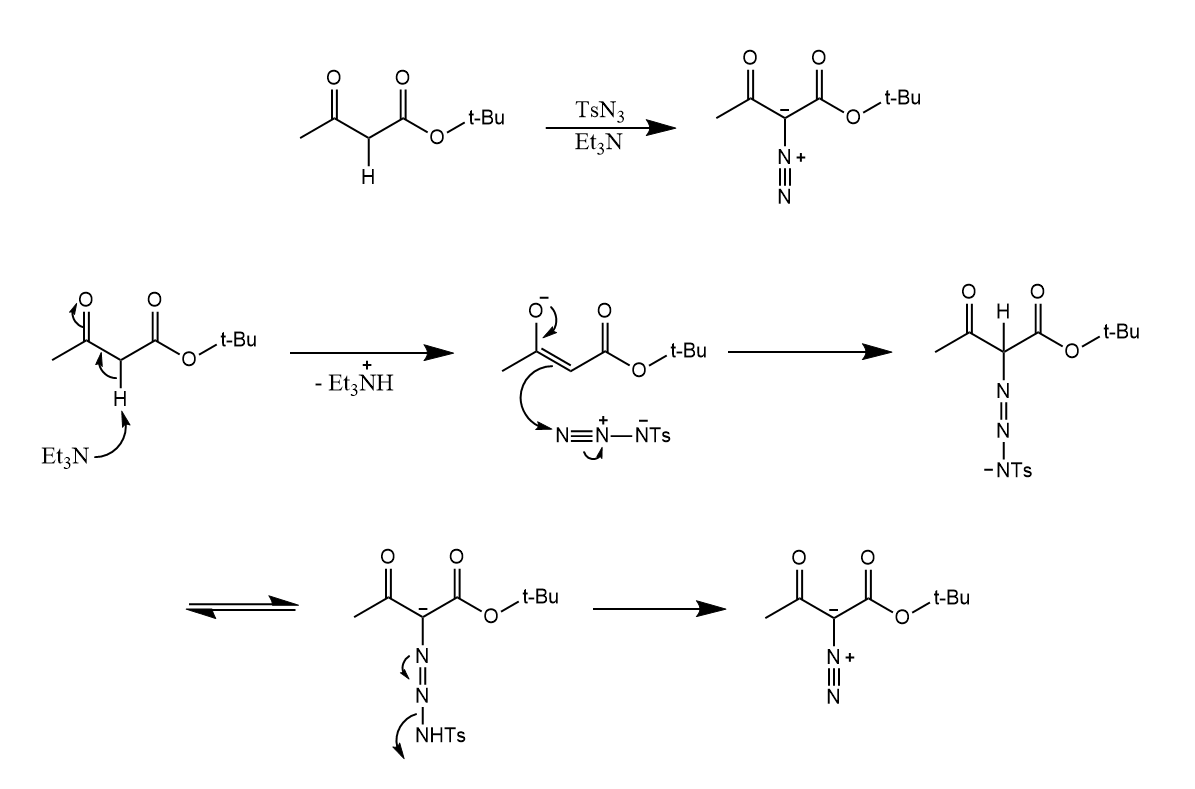
而重氮化合物又可通过磺酰腙在碱性条件下反应得到,或通过酰氯和重氮甲烷反应得到α-重氮羰基化合物,或使用Regitz重氮转移:
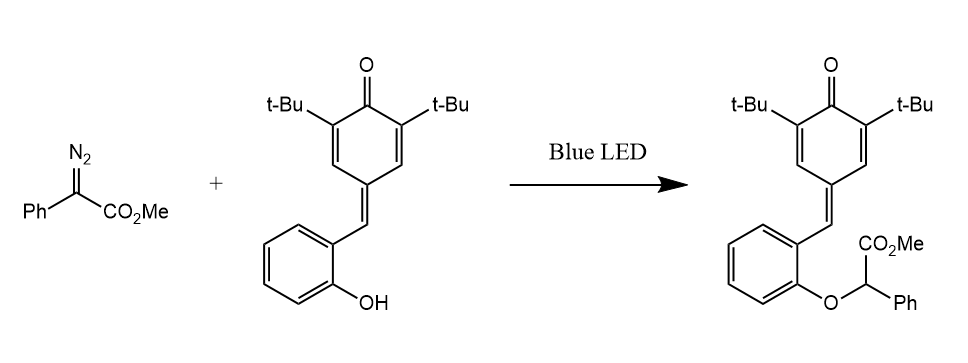
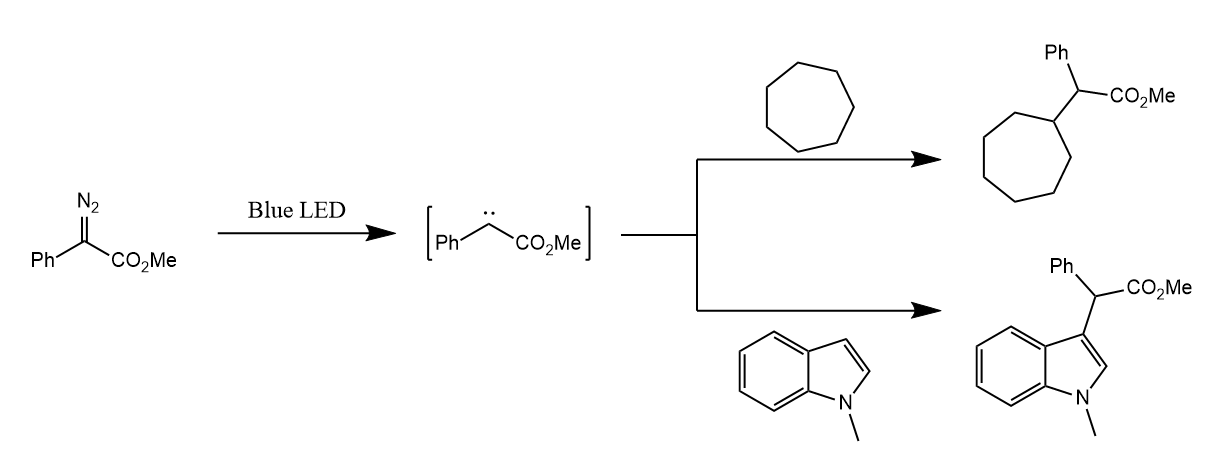
由重氮化合物光解形成的卡宾可发生O-H、S-H、N-H和C-H键等的插入反应。
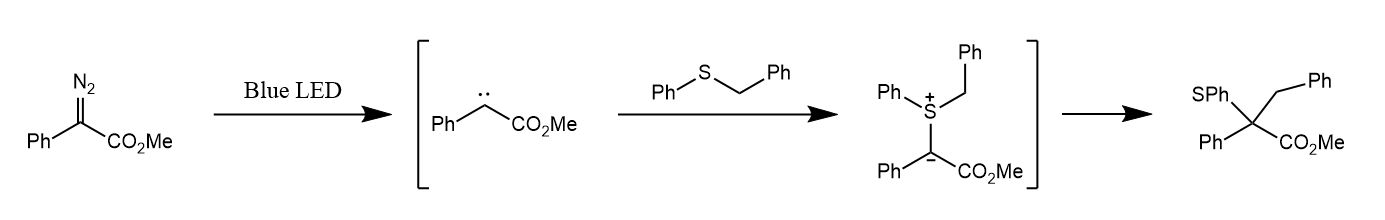
卡宾的空p轨道可以接受硫醚中硫原子的孤对电子,形成锍盐中间体,继而发生[1,2]-σ迁移得到Stevens重排产物。
若原料是烯丙基硫醚,则发生[2,3]-σ迁移得到Doyle-Kirmse重排产物。
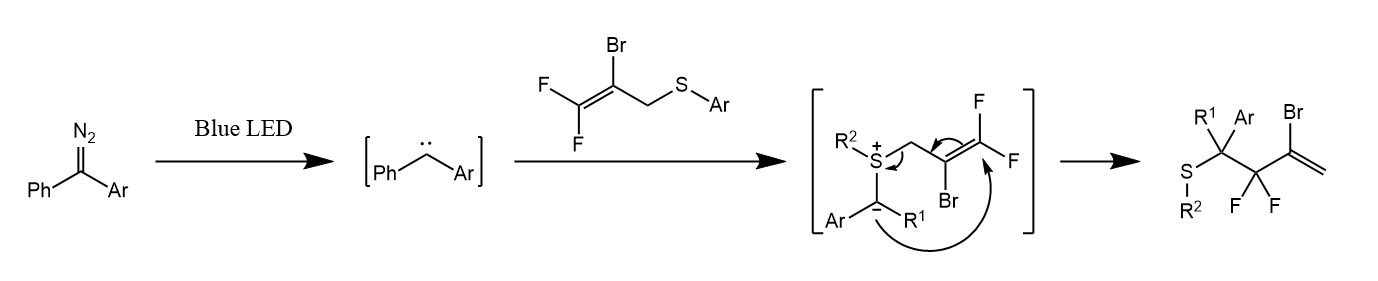
卡宾能与烯烃发生环丙烷化反应,例如,由α-重氮酯热分解所形成的卡宾被苯乙烯捕获,生成环丙烷化产物。
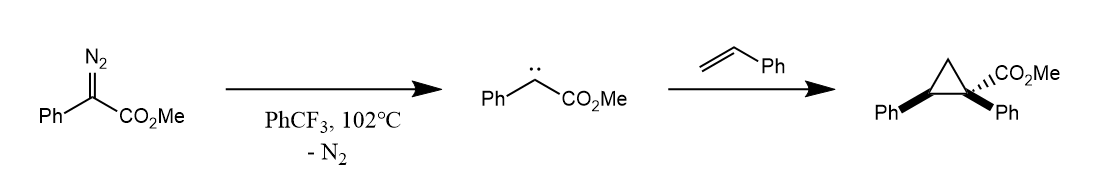
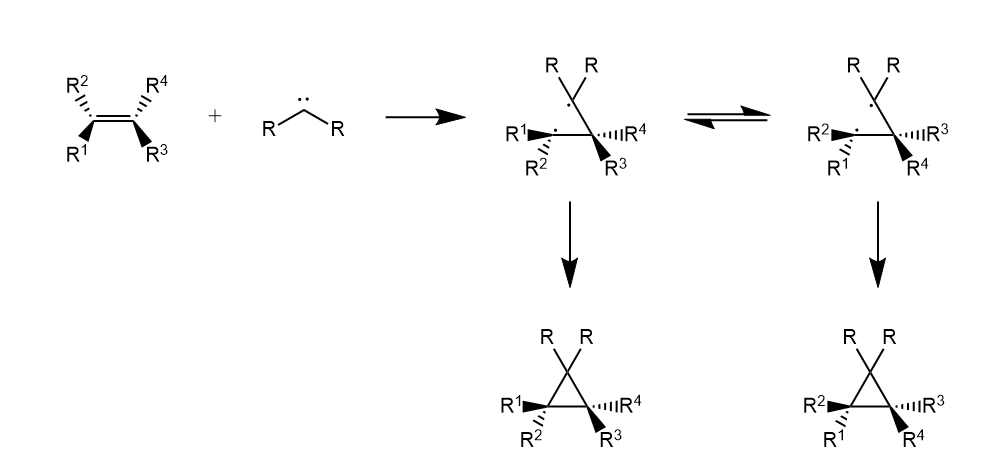
卡宾的环丙烷化通过一个协同的过程,立体专一性地生成多取代的环丙烷衍生物,烯烃的构型在环丙烷化过程中得以保留,卡宾上两个取代基的位置则取决于基团体积大小。
至于三线态卡宾,可以看作是双自由基,经历分布的自由基加成机理,故立体选择性较差。
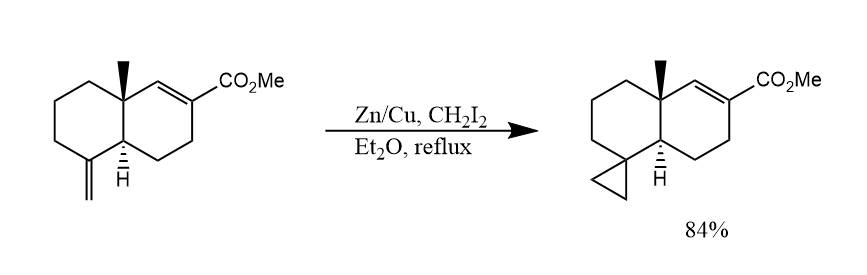
二碘甲烷和Zn(Cu)生成的试剂$ICH_2ZnI$,称为Simmons-Smith试剂,这是一种类卡宾试剂,可以像卡宾一样与烯烃发生环丙烷化。
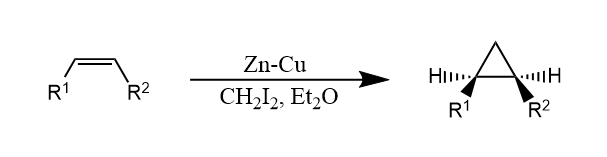
另一种类似的试剂是$Et_2Zn$和$CH_2I_2$生成的Furukawa试剂,也广泛被用在Simmons-Smith环丙烷化中,反应具有立体专一性。
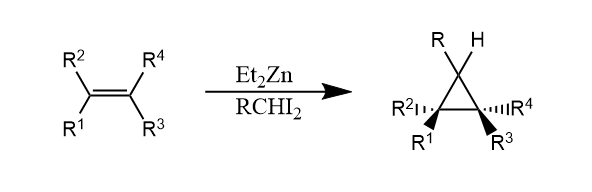
上述两种金属卡宾都是缺电子性的,因此它和富电子烯烃的反应性更优。
卡宾虽然是缺电子性的,但是也能被用于亲核取代或亲核性催化剂,典型例子如氮杂化卡宾(NHC)替代氰基催化的安息香缩合。
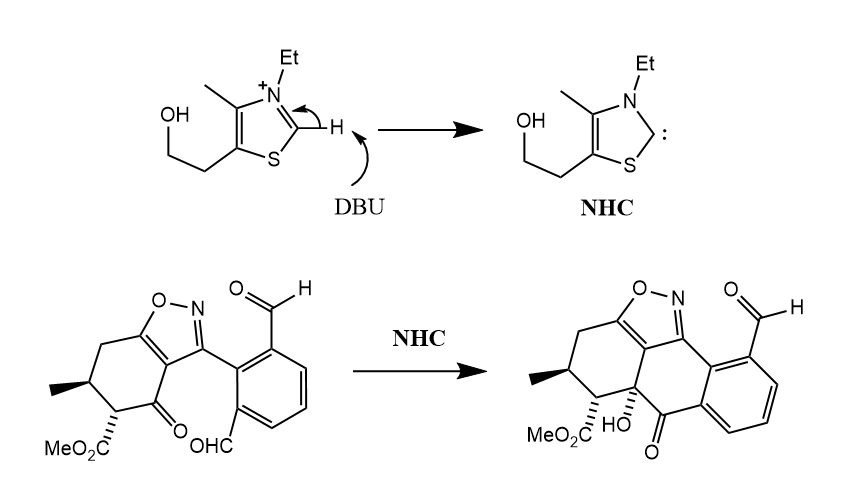
( 这个机理太难画了,我就不画了XD,反应本身就是常见的安息香缩合,常规的安息香缩合用的是氰基催化,但是考虑到氰化物的毒性,所以NHC作为其替代是一个很好的选择。 )
10.5 亚基卡宾的形成及其反应
如果六电子的卡宾连有双键,那么称为亚基卡宾(alkenylidene)。亚基卡宾和三键之间可以通过[1,2]-迁移进行互变。
碱性条件下,α-重氮膦酸酯和羰基化合物反应可以形成亚基卡宾中间体。
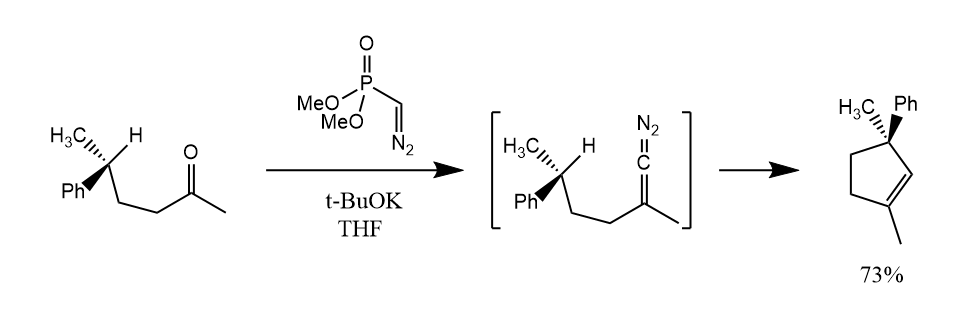
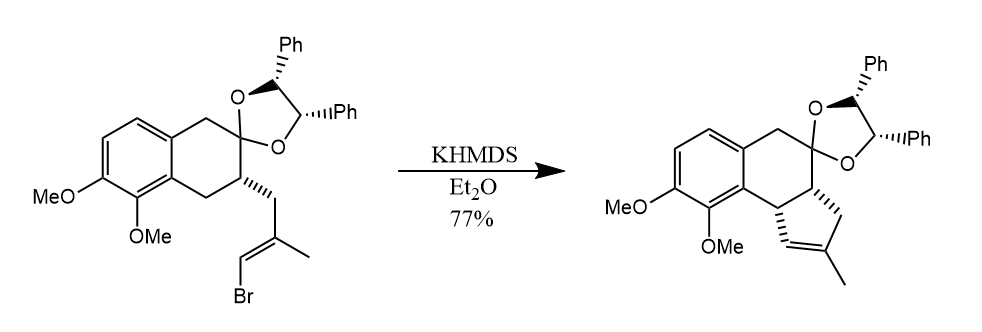
亚基卡宾中间体也可以通过α-消除反应产生,在碱性条件下,溴代烯烃发生α-消除得到亚基卡宾,继而被分子内C-H键捕获得到稠环产物。
10.6 乃春的形成及其反应
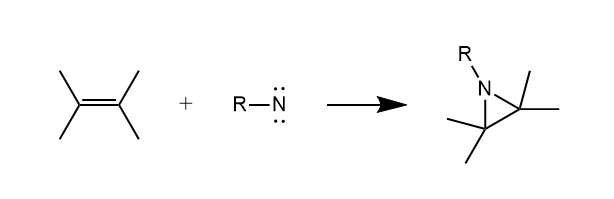
与卡宾类似,乃春能对碳碳重键发生加成,生成氮杂环丙烷,又称为吖丙啶。
乃春可由叠氮化合物的热解或光解产生。
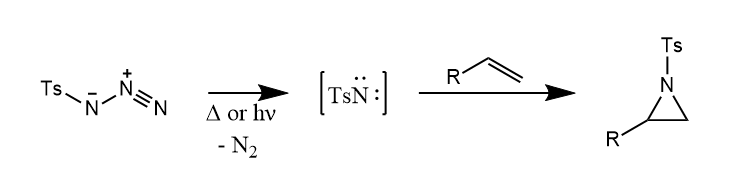
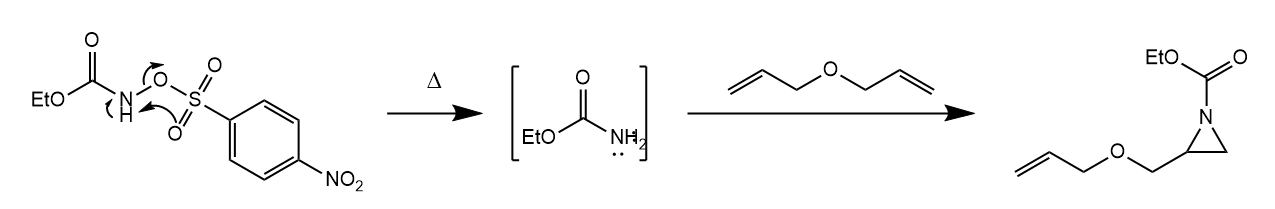
氮原子上连有离去基团的胺或酰胺亦可经α-消除形成乃春。
总结
本章讨论了自由基反应和卡宾/乃春等反应,自由基反应是基础有机中讨论较少的部分,所以本章的介绍写的更详细一点点。
我在基础有机中学完自由基反应后的印象就是反应难以控制,选择性较差等等。但是随着光化学、过渡金属催化等等领域的不断发展,上述缺点不断被克服,自由基反应也越来越常见和越来越重要,许多更前沿的内容在本书中介绍的不多,有待更进一步了解。