《中级有机化学》章末总结7
五一假期写一下第七章总结,第七章为亲电取代反应。
第7章 亲电取代反应
7.1 芳环上的亲电取代反应
芳环上的氢被亲电试剂所取代,称为芳香烃亲电取代反应($S_EAr$)。
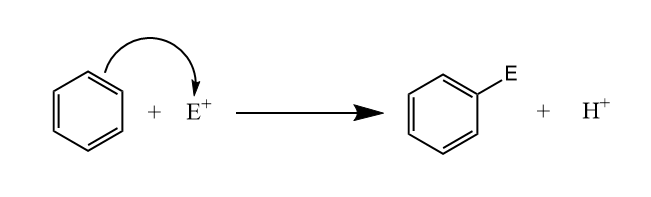
芳环上亲电取代反应包括以下两步:第一步为芳环对亲电试剂的加成,生成σ-络合物;第二步为$\beta-H$消除,其中第一步为决速步。
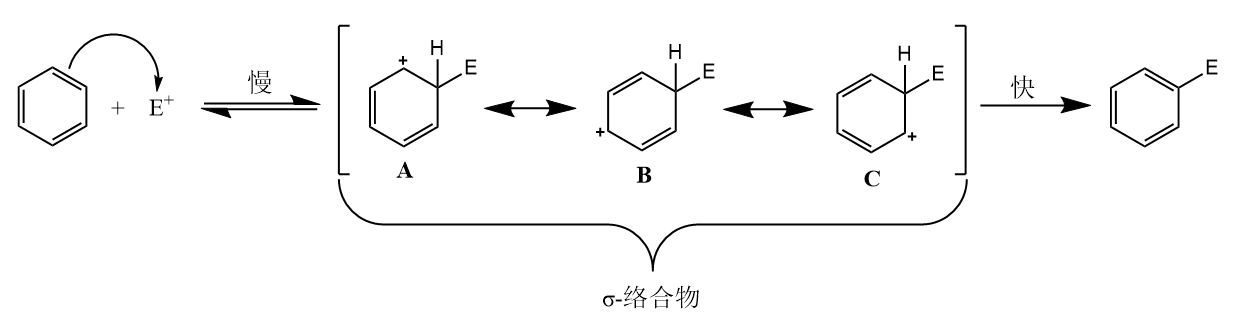
σ-络合物的生成已经实验证明,一些相对稳定的络合物中间体可以在低温下稳定存在并被检测出来。
芳香亲电取代反应根据亲电试剂的不同分为卤化、硝化、磺化、烷基化和酰基化等。这些部分在基础有机中已经有详细的论述,下文只选择性地介绍部分内容。
卤代烃在Lewis酸作用下,$C-X$键发生极化生成碳正离子,碳正离子作为亲电试剂接受芳烃进攻的反应称为Friedel-Crafts烷基化反应(简称F-C烷基化)。
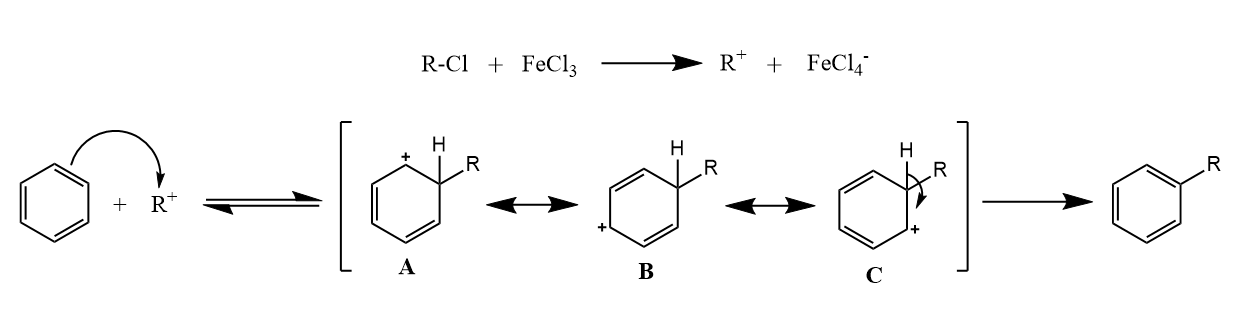
F-C烷基化有两个缺点:由于烷基的给电子性,反应往往会得到多取代苯;烷基碳正离子的生成可能会导致重排,使产物复杂化。
F-C烷基化反应是可逆的,下面有一个有趣的反应可以体现这一点:
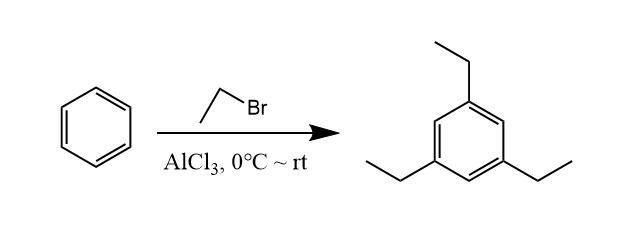
乙基是邻对位定位基,但是上述反应却得到了均三乙苯。这是因为反应生成的邻对位烷基化产物位阻大,由于反应的可逆性发生烷基化的逆反应并再次烷基化,生成热力学稳定的均三乙苯。
甲醛和盐酸在氯化锌催化下与芳烃反应生成氯甲基化的芳烃产物的反应叫做氯甲基化反应。
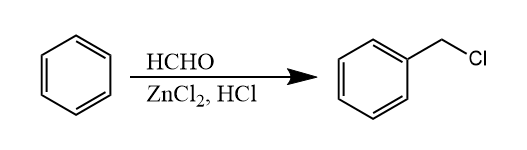
芳香烃在Lewis酸催化下与酰氯或酸酐反应生成酰基化产物,称为Friedel-Crafts酰基化反应(简称F-C酰基化)。
F-C酰基化与烷基化相比,酰基化生成的产物活性较低,一般不发生多酰基化;同时酰基化反应不生成烷基碳正离子,因此不会发生重排。
芳环上亲电取代反应的反应活性取决于芳环上的电子密度,因此给电子基会活化芳环,吸电子基则会钝化芳环。芳杂环的电子密度受杂原子孤对电子的取向影响,例如吡咯的亲电取代活性大于苯大于吡啶。
芳环上亲电取代反应的区域选择性取决于芳环上取代基的定位效应,取代基一般分为邻对位取代基和间位取代基两类。
对于反应活性和区域选择性,本文不多介绍,可参阅基础有机化学教材。
下面介绍几个芳环上亲电取代的相关反应。
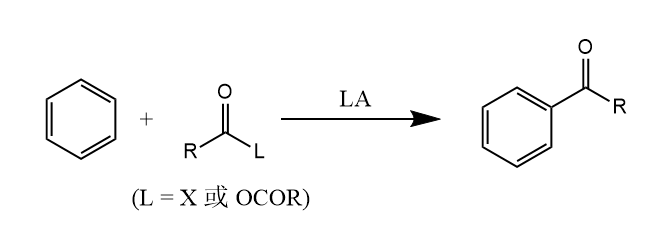
一氧化碳和$HCl$原位产生的酰化试剂可与芳烃产生酰基化反应,生成芳甲醛,该反应称为Gattermann-Koch甲酰化反应,这个反应一般只适合苯和烷基苯。
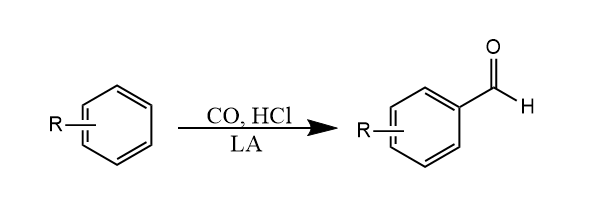
与Gattermann-Koch甲酰化反应相似,当酰化试剂使用$HCN/HCl$时,反应得到甲酰化产物,称为Gattermann甲酰化,这个反应适合烷基苯、苯酚和烷氧基苯。
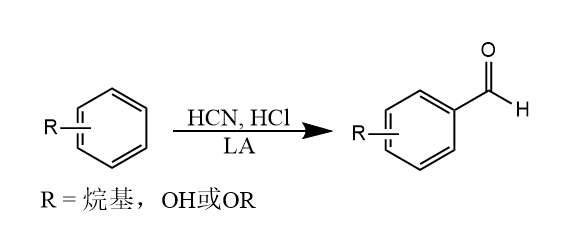
作为Gattermann反应的一个扩展,用腈代替$HCN$作为酰基化试剂,可在质子酸/Lewis酸存在下由芳烃制备为酮。
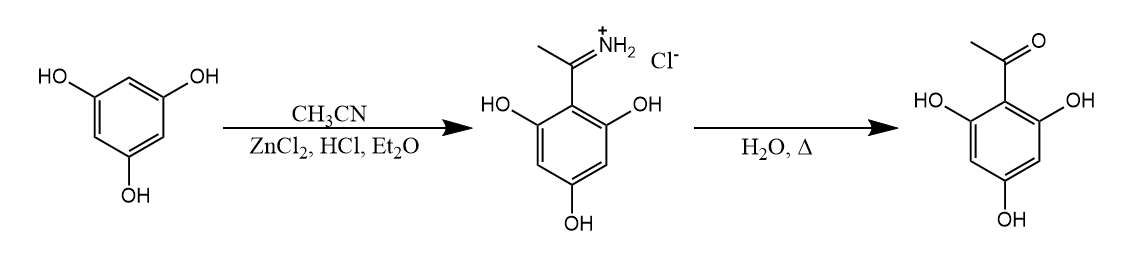
在三氯氧膦存在下,DMF作为酰基化试剂与富电子性芳香烃发生甲酰化反应称为Vilsmeier-Haack甲酰化反应。
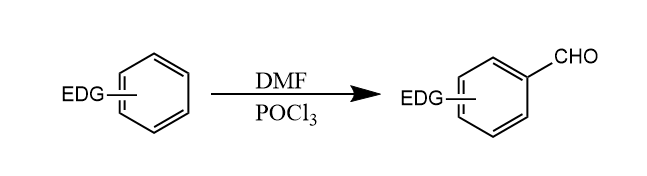
该反应经历氯代亚胺盐的亲电中间体。
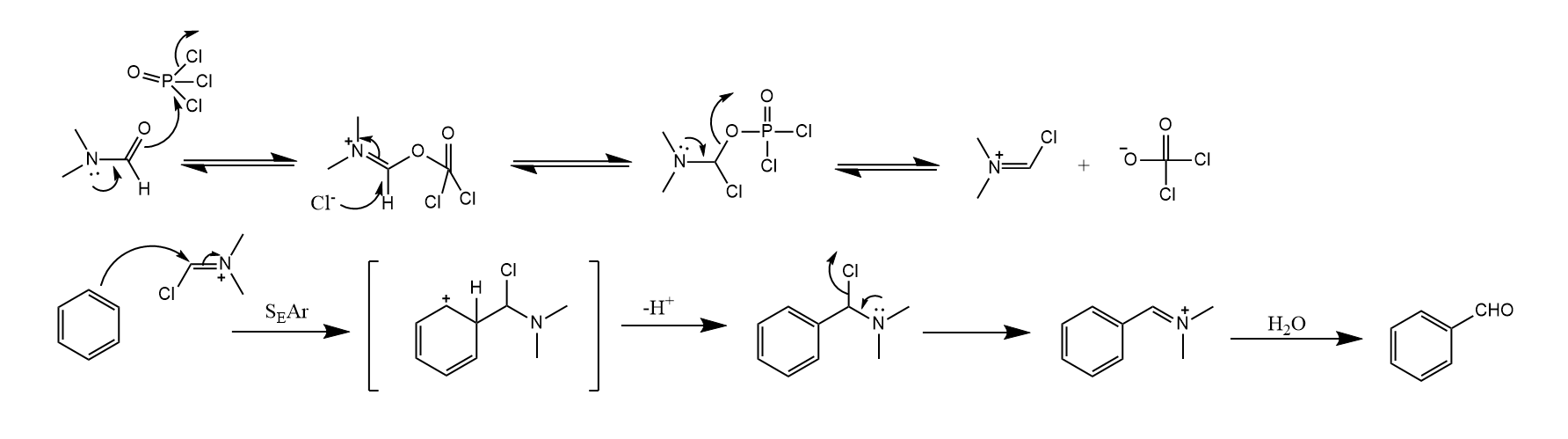
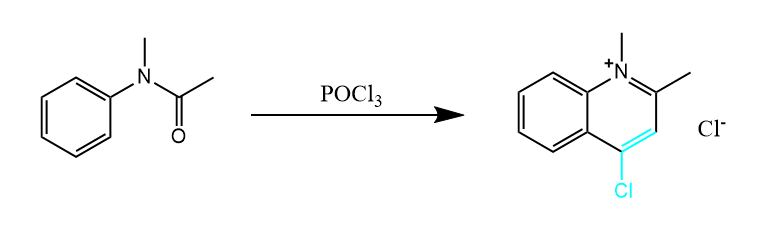
Vilsmeier最初发现这一反应时,用的底物为N-甲基乙酰苯胺,该反应中涉及两分子底物的反应。
当富电子性芳烃用氯仿和氢氧化钠处理时,得到芳香烃甲酰化产物,该反应称为Reimer-Tiemann反应。
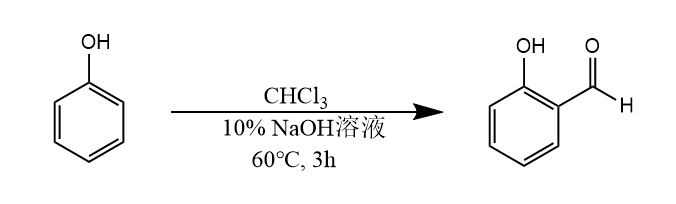
该反应须在强碱条件下进行,反应过程涉及二氯卡宾中间体。
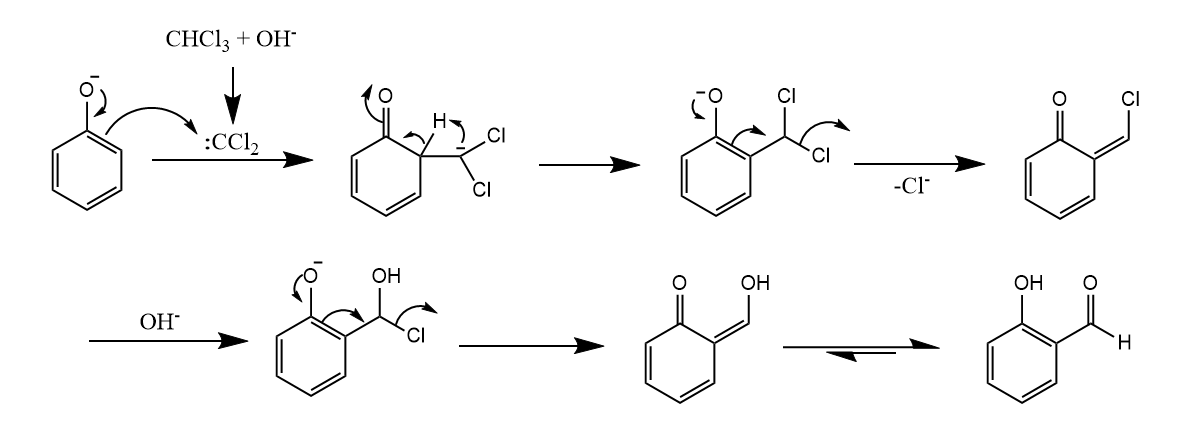
苯酚的羧酸酯在Lewis酸或质子酸催化下重排成邻位或对位酰基酚的反应称为Fries重排。
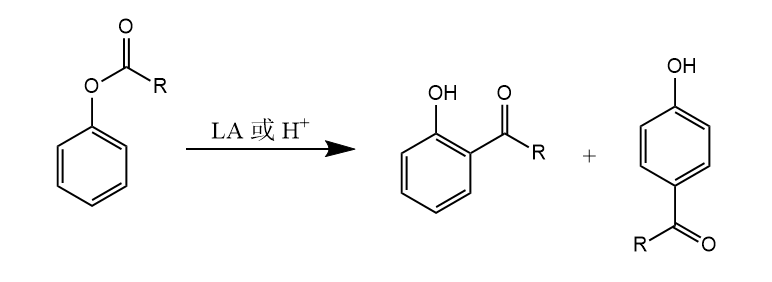
两个芳基在Lewis酸和质子酸存在下,先生成欧联中间体,继而脱氢芳构化,该反应称为Scholl反应。
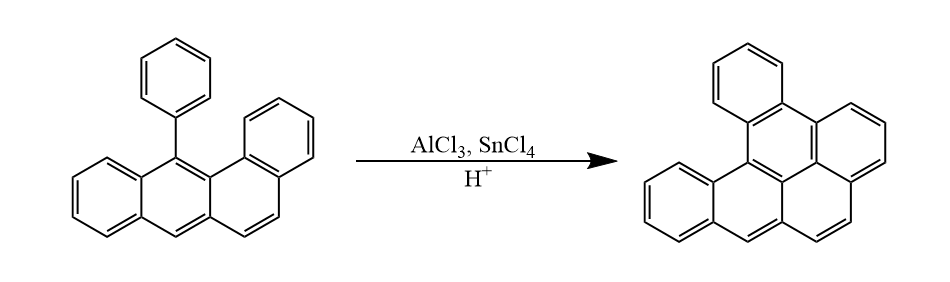
Scholl反应易在分子内发生,分子间的Scholl反应需要脱氢试剂帮助完成芳构化,如二氰基二氯苯醌(DDQ)等。
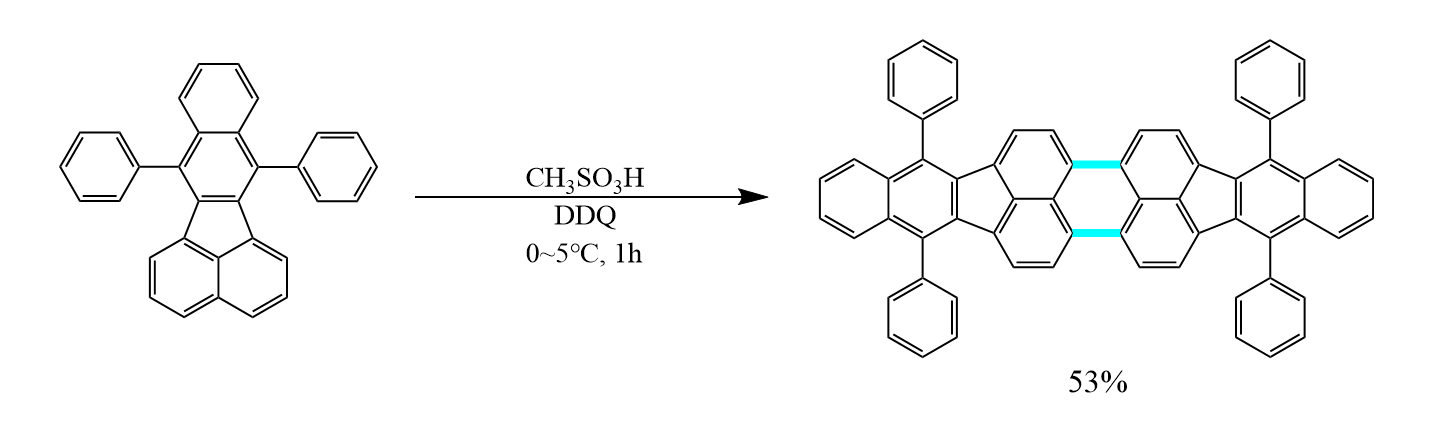
一些芳香烃的亲电取代反应是可逆的,这些逆的亲电取代反应称为同位(ipso)亲电取代反应。一个常见的例子是磺化反应的逆反应,即苯磺酸脱磺基的过程就是芳香同位亲电取代反应。
另一个例子是2-苯甲酰基-3-甲基苯甲酸在酸性条件下的重排:
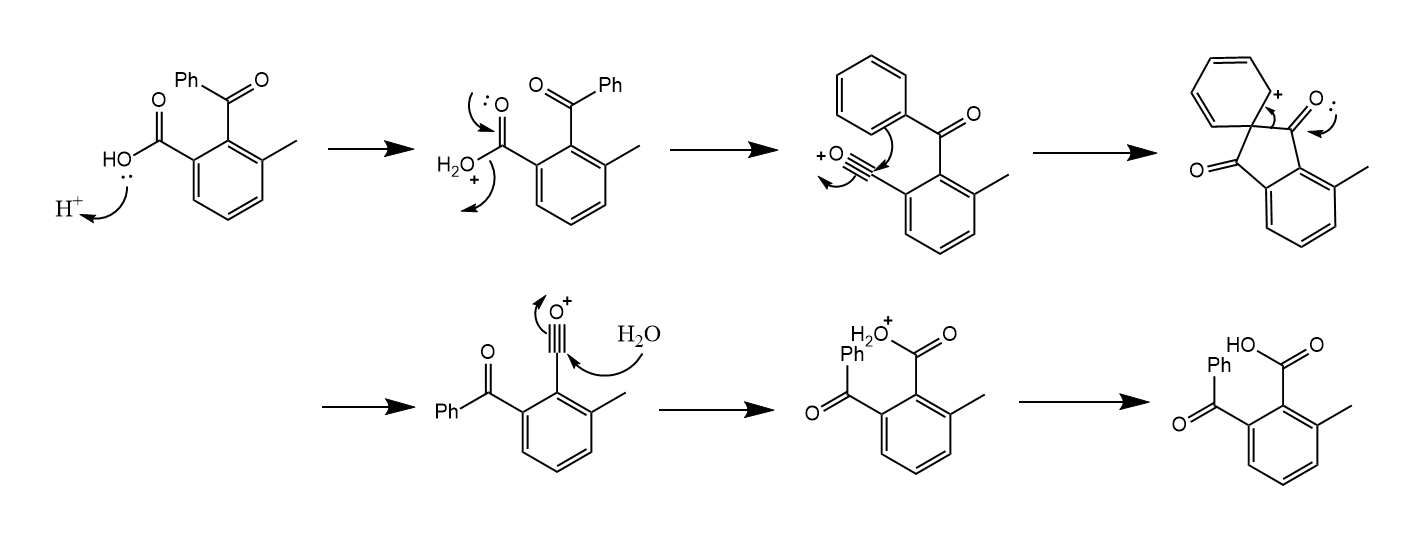
反应通过同位Friedel-Crafts酰基化实现重排。
7.2 羰基化合物α-碳上的亲电取代反应
含有α-H的羰基化合物可以在碱性或酸性条件下发生酮式-烯醇互变异构。在碱性条件下,碱夺得羰基化合物的α-H形成碳负离子,继而共振为烯醇负离子,获得质子后成烯醇式;在酸性条件下,质子化的羰基化合物共振为碳正离子,β-H消除形成烯醇式。
影响酮式和烯醇式分布的主要因素有三:α-H酸性、体系酸碱性和溶剂。前两点易于理解,氢酸性越强,体系碱性越强,越易生成烯醇式;非质子溶剂易于生成烯醇式,因为烯醇式可能存在分子内氢键或分子间氢键,在非质子溶剂中更为稳定。
羰基化合物的α-卤化在酸性和碱性条件均能发生,机理在基础有机中均已介绍,此处只简要加以说明。
碱性条件下,甲基酮与卤素反应生成羧酸和卤仿,称为卤仿反应(haloform reaction)。
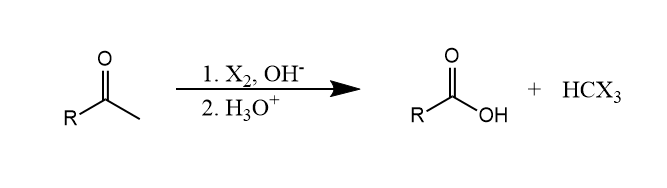
卤仿反应说明了碱性条件下的α-卤代往往会发生多取代,且产物为动力学产物,因为卤素的吸电子诱导效应会更利于负离子中间体的形成。
酸性条件下的α-卤代反应具有更好的可控性,反应停在单卤代热力学产物,若亲电试剂过量,则生成1,3-二卤代产物。
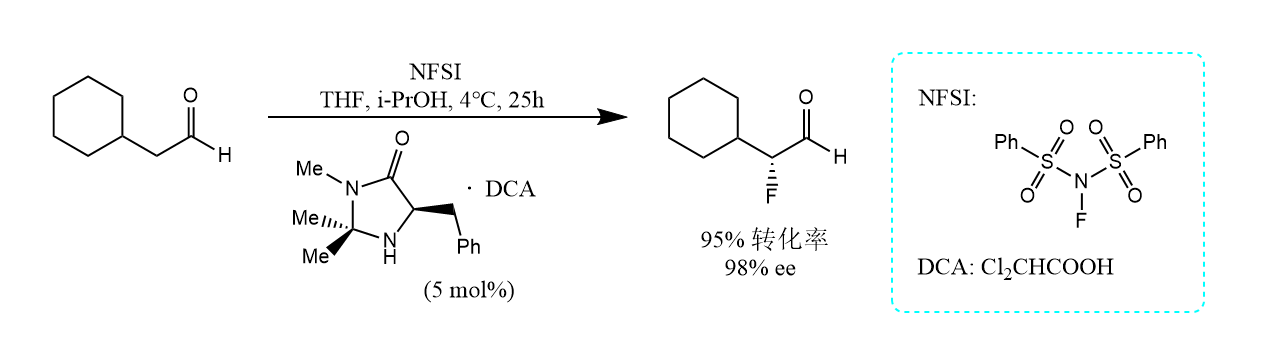
醛不能直接卤化,因为醛易被氧化为酸,想将醛α-卤代可将醛先转化为缩醛或烯胺,卤化后再水解,即可间接得到α-卤代醛。例如借助手性胺实现醛的不对称α-氟化反应:
羧酸α-H的酸性比醛酮羰基α-H的酸性弱,所以羧酸的α-卤代需要将羧酸先转化为酰氯或酸酐。一个经典反应是Hell-Volhard-Zelinsky反应(HVZ反应)。
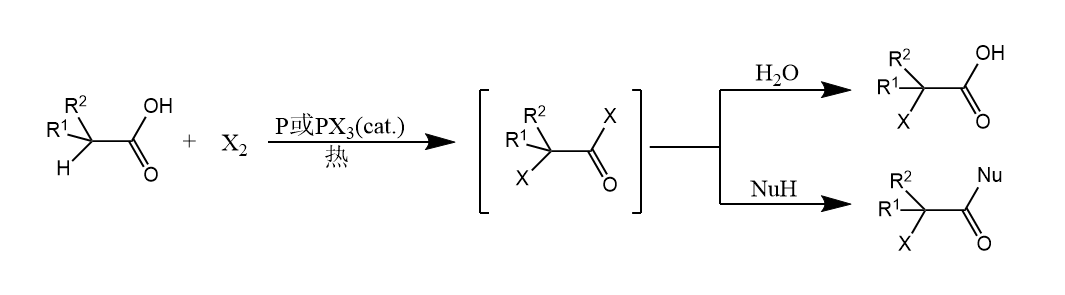
羧酸卤化的试剂对象是酰卤,完成卤化后酰卤水解得到α-卤代羧酸。
羰基α-H被碱夺取后,形成的烯醇负离子进攻卤代烷或磺酸酯等亲电试剂,生成α-烷基化产物,称为羰基化合物的α-烷基化反应。α-烷基化也可以称作C-烷基化,考虑到烯醇负离子是两可亲核试剂,所以烯醇的氧也可能会进攻亲电试剂,生成O-烷基化产物,这两个反应存在相互竞争。
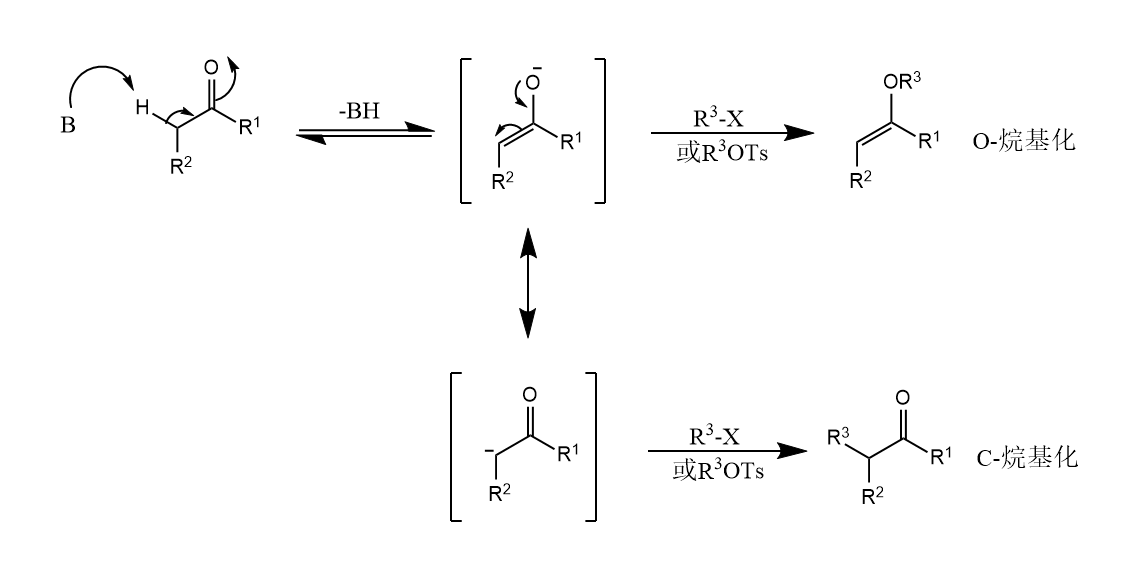
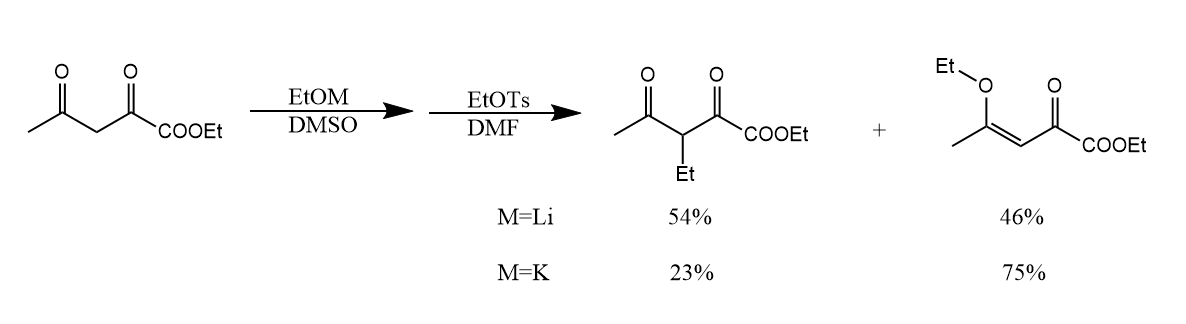
因此提高C-烷基化和O-烷基化的选择性就成为了一个关键的问题,使用的碱和溶剂有时会影响亲电的选择性。例如使用$EtOLi$或$EtOK$催化反应,形成的烯醇负离子的氧与金属结合,其中$O-Li$键共价成分多,而$O-K$键具有更多离子键特征,且$K^+$易被DMSO络合稳定,所以使用$EtOK$作为碱易生成O-烷基化产物。
提高选择性的另一种思路是让烯醇式的氧失去亲电能力,例如通过烯醇硅醚/烯胺实现α-烷基化。
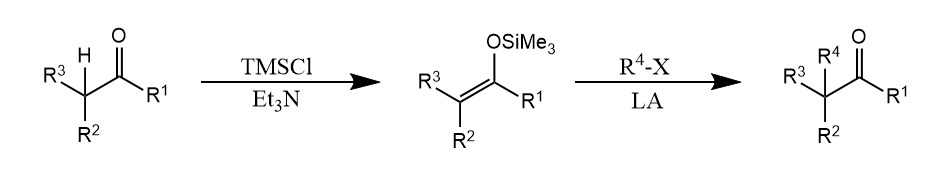
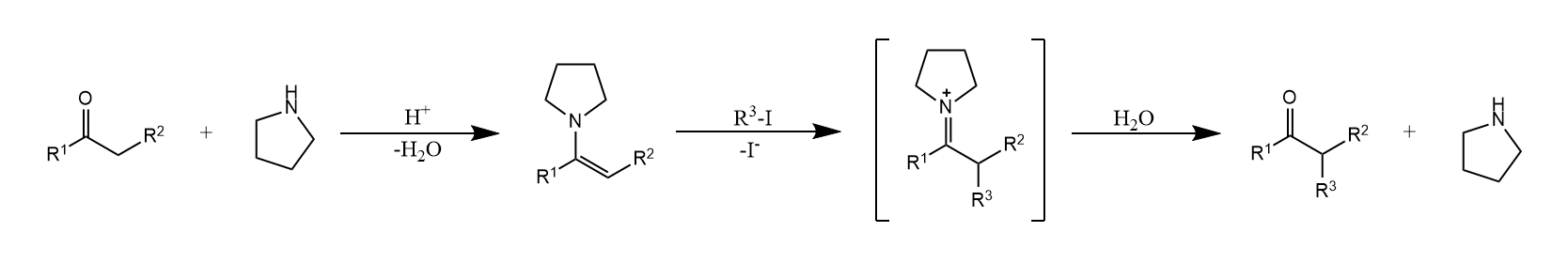
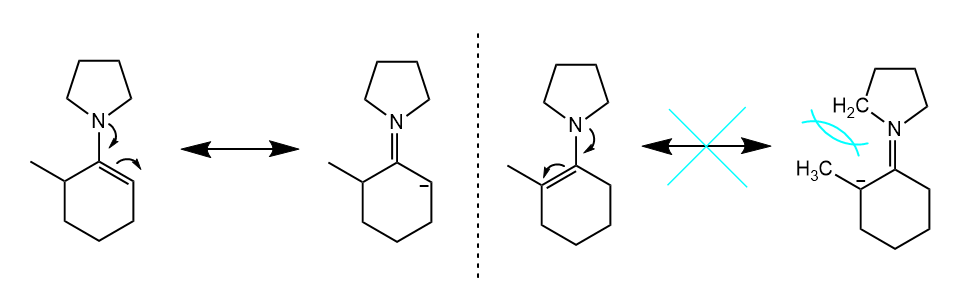
使用烯醇硅醚和烯胺这两种方法的不同之处在于,羰基化合物往往倾向生成热力学烯醇硅醚产物,而生成动力学烯胺产物,其原因是热力学烯胺的位阻往往较大,影响烯胺共振。
在α-烷基化过程中也要考虑反应的区域选择性,例如下面的例子:
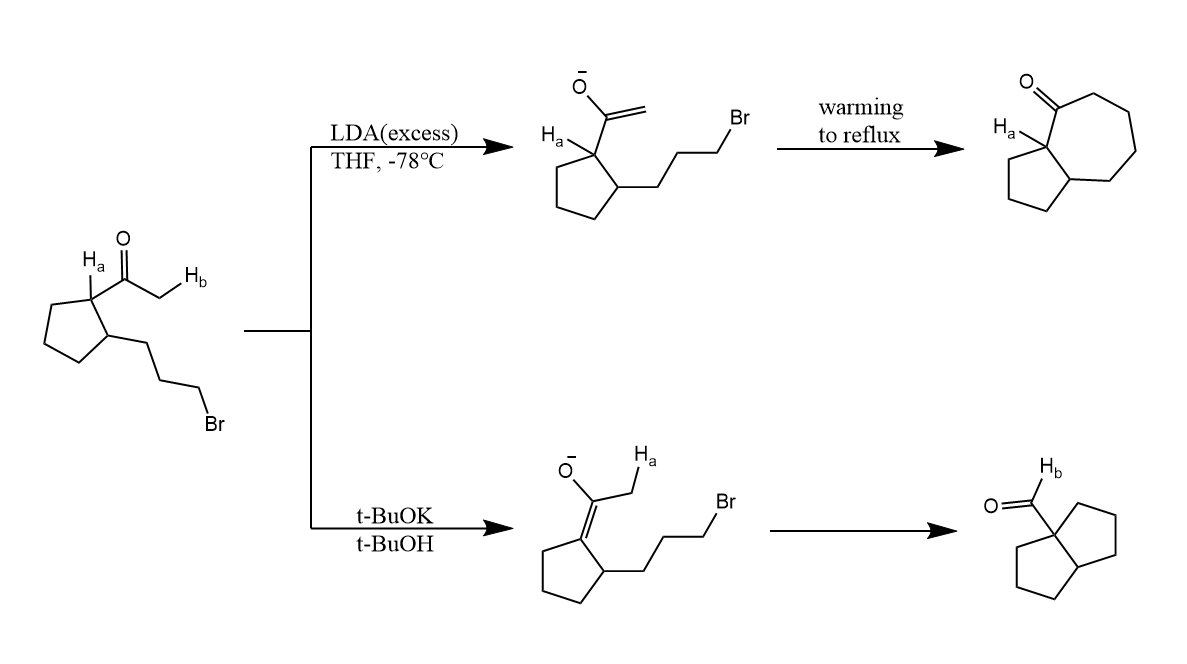
低温+大位阻碱易生成动力学产物,反之则容易生成热力学产物。
羰基化合物的α-烷基化是合成中十分常见的手段,其中乙酰乙酸乙酯和丙二酸二乙酯是两个非常常用的原料,它们的合成原理也十分相似,都是先烷基化,然后水解脱羧,得到α-烷基化乙酮/乙酸。详细的原理已在基础有机化学中涉及,此处略去。
除了烷基化,还有α-酰基化,羧酸酯在碱性条件下的α-酰基化反应称为Claisen酯缩合。
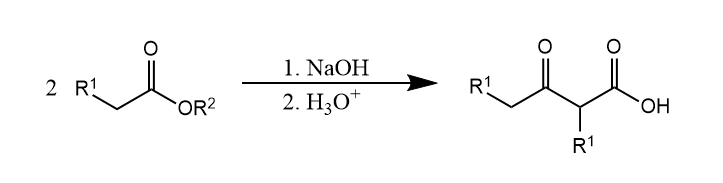
该反应也能在分子内进行,分子内的酯缩合也称Dieckmann缩合。
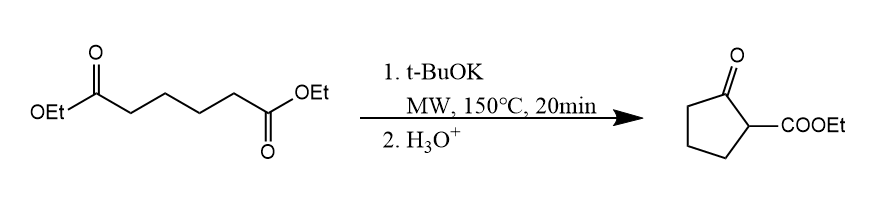
两反应的机理都是类似的,酯形成烯醇负离子,然后亲核加成另一分子(或自身)的羰基,得到β-酮酸酯产物。机理在此略去。
如果是两个不同的酯发生缩合时,可能会交叉缩合得到复杂产物,所以往往将一分子反应物选择为无α-H的酯,例如:
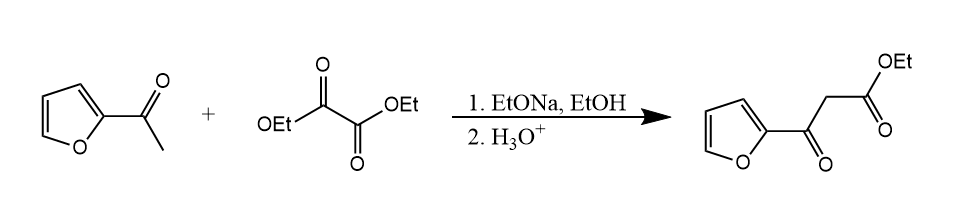
最后提一下α-重氮反应,在碱性条件下,含两个α-H的羰基化合物和$TsN_3$反应,生成α-重氮羰基化合物,尤其适合环状重氮化合物的生成。
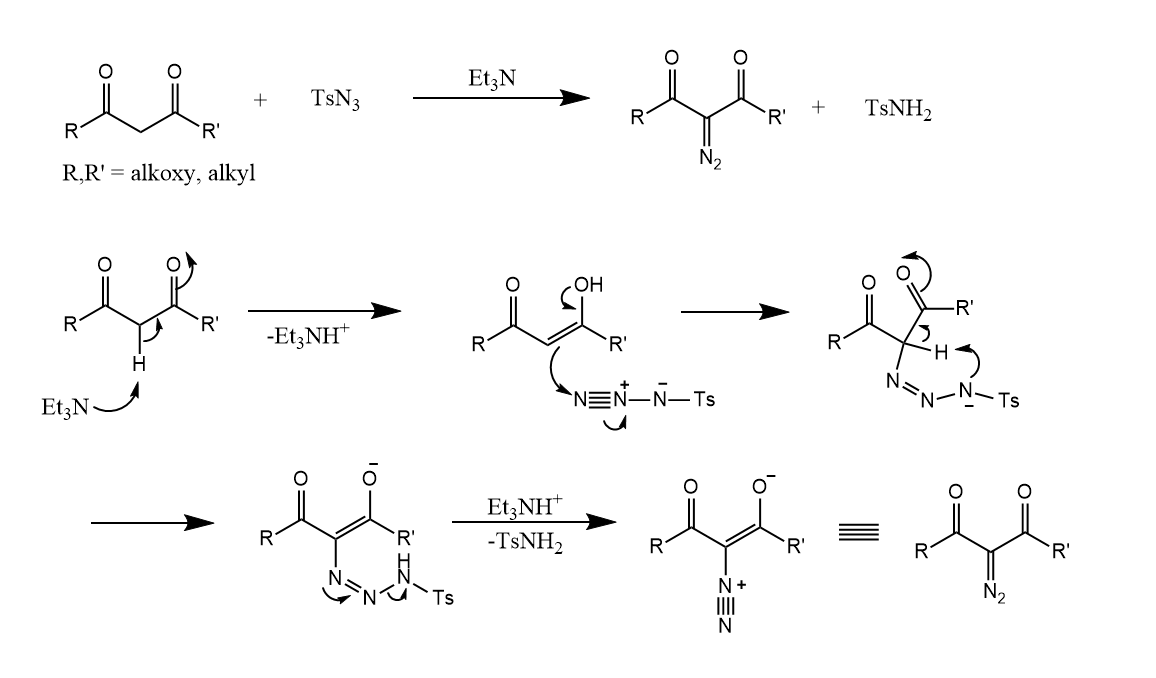
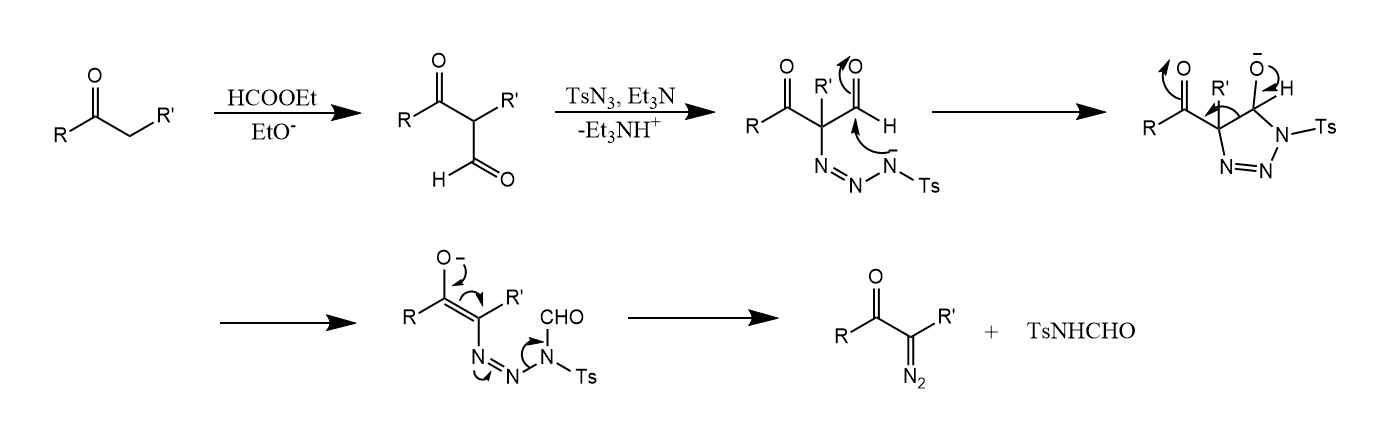
由于一般羰基化合物的α-酸性不够强,所以该反应往往和Claisen缩合联用,对底物进行活化。
总结
本章基本是复习了基础有机的内容,没什么新东西。
画图ing