8.1 The Variational Method Provides an Upper Bound to the Ground-State Energy of a System
We will first illustrate the variational method. Consider the ground state of some arbitrary system. The ground-state wave function ψ0 and energy E0 satisfy the Schrödinger equation:
H^ψ0=E0ψ0
We can obtain:
E0=⟨ψ0∣ψ0⟩⟨ψ0∣H^∣ψ0⟩
A beautiful theorem says that if we substitute any other function ϕ for ψ0 into the equation and calculate the corresponding energy:
Eϕ=⟨ϕ∣ϕ⟩⟨ϕ∣H^∣ϕ⟩
then Eϕ will be greater than the ground-state energy E0. In an equation, we have the variational principle:
Eϕ≥E0
where the equality holds only if ϕ=ψ0, the exact wave function.
The variational principle says that we can calculate an upper bound to E0 by using any trial function we wish. The closer ϕ is to ψ0 in some sense, the closer Eϕ will be to E0.
We can choose a trial function ϕ such that it depends upon some arbitrary
parameters, α,β,γ,⋯, called variational parameters. The energy also will depend upon these variational parameters:
Eϕ(α,β,γ,⋯)≥E0
Now we can minimize Eϕ with respect to each of the variational parameters and thus approach the exact ground-state energy E0.
The Schrödinger equation cannot be solved exactly for this system because of the term involving r12.
We can rewrite the equation and obtain:
H^=H^H(1)+H^H(2)+4πϵ0e2r121
where:
H^H(j)=−2meℏ2∇j2−4πϵ02e2rj1j=1and2
is the Hamiltonian operator for a single electron around a helium nucleus. If we ignore the interelectronic repulsion term, then the Hamiltonian operator is separable and the ground-state wave function would be:
ϕ0(r1,r2)=ψ1s(r1)ψ1s(r2)
where:
ψ1s(rj)=(πa0Z3)1/2e−Zrj/a0j=1or2
where a0=4πϵ0ℏ2/mee2. We use these equation as a trial function using Z as a variational constant. Thus, we must evaluate:
E(Z)=∫ϕ0(r1,r2)H^ϕ0(r1,r2)dr1dr2
The integral is a bit lengthy, we can get the results:
E(Z)=16π2ϵ02ℏ2mee4(Z2−827Z)
It is convenient to express E in units of mee4/16π2ϵ02ℏ2. If we minimize E(Z) with respect to Z, we find that Zmin=27/16. So we get:
Emin=−(1627)2=−2.8477
which is in good agreement with the experimental result of −2.9033. Thus, we achieve a fairly good result, considering the simplicity of the trial function.
8.2 A Trial Function That Depends Linearly on the Variational Parameters Leads to a Secular Determinant
As another example of the variational method, consider a particle in a one-dimensional box. we should expect it to be symmetric about x=a/2 and to go to zero at the walls. One of the simplest functions with these properties is xn(a−x)n, where n is an integer. Consequently, let’s estimate E0 by using:
ϕ=c1x(a−x)+c2x2(a−x)2
as a trial function, where c1 and c2 are to be determined variationally—that is, where c1 and c2 are the variational parameters. With this trial function, we can get the result:
Emin=0.125002ma2h2
compared with:
Eexact=0.125000ma2h2
So we see that using a trial function with more than one parameter can produce impressive results. The price we pay is a correspondingly more lengthy calculation.
Fortunately, there is a systematic way to handle a trial function, which can be written generally as:
Thus, we obtain a secular determinant and a secular equation.
The quadratic secular equation gives two values for E, and we take the smaller of the two as our variational approximation for the ground-state energy.
The excellent agreement here (remember Eexact=0.125000mh2) is better than should be expected normally for such a simple trial function.
We can also determine the normalized trial function for our variational treatment of a particle in a box:
c1c2=−H12−ES12H11−ES11=1.13342
ϕ(x)=c1[x(1−x)+1.13342x2(1−x)2]
∫01ϕ2(x)dx=c12∫01[x(1−x)+1.13342x2(1−x)2]2dx=1
So we can obtain:
ϕ(x)=4.40378x(1−x)+4.99133x2(1−x)2
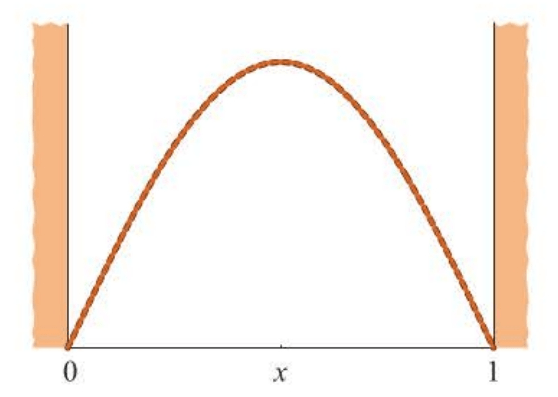
Figure 8.1 is a comparison of the optimized and normalized trial function with the exact ground-state particle-in-a-box wave function, ψ1(x)=2sinπx. The two curves are essentially the same.
If we use a linear combination of N functions, instead of using a linear combination of two functions as we have done so far, then we obtain N simultaneous linear algebraic equations for the cj's. We can express compactly by using the matrix notation:
Hc=ESc
where H is an N×N matrix with matrix elements Hij, S is an N×N matrix with matrix elements Sij, and c is an N×1 column matrix whose elements are cj.
To have a nontrivial solution to this set of homogeneous equations, we must have:
∣H−ES∣=0
The determination of the smallest root must usually be done numerically for values of N larger than two. This is actually a standard numerical problem, and a number of packaged computer programs do this.
8.3 Trial Functions Can Be Linear Combinations of Functions That Also Contain Variational Parameters
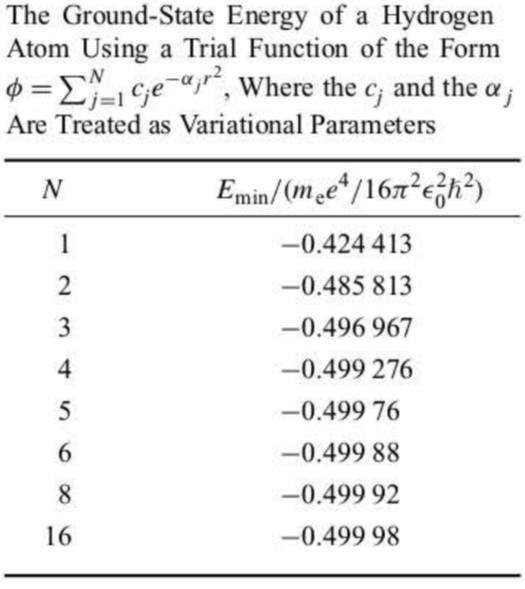
An example of a trial function for the hydrogen atom is:
ϕ=j=1∑Ncje−αjr2
where the cj's and the αj's are treated as variational parameters. ϕ is linear only in the cj but not in the αj. The minimization of E with respect to the cj and αj is fairly complicated, involving 2N parameters, and must be done numerically.
8.4 Perturbation Theory Expresses the Solution to One Problem in Terms of Another Problem That Has Been Solved Previously
Suppose we wish to solve the Schrödinger equation for some particular system, but we are unable to find an exact solution as we have done for a harmonic oscillator, a rigid rotator, and a hydrogen atom in previous chapters. It turns out that most systems cannot be solved exactly; some specific examples are a helium atom, an anharmonic oscillator, and a nonrigid rotator.
For example, the Hamiltonain operator for a helium atom is:
Another example of a problem that could be solved readily if it were not for additional terms in the Hamiltonian operator is an anharmonic oscillator.
H^=−2μℏ2dx2d2+21kx2+61γ3x3+241γ4x4
These two examples, with their Hamiltonian operators, introduce us to the basic idea behind perturbation theory.
In both of these cases, the total Hamiltonian operator consists of two parts, one for which the Schrödinger equation can be solved exactly and an additional term, whose presence prevents an exact solution. We call the first term the unperturbed Hamiltonian operator and the additional term
the perturbation.
We shall denote the unperturbed Hamiltonian operator by H^(0) and the perturbation by H^(1) and write:
H^=H^(0)+H^(1)
Associated with H^(0) is a Schrödinger equation, which we know how to solve, and so we have:
H^(0)ψ(0)=E(0)ψ(0)
where ψ(0) and E(0) are the known eigenfunctions and eigenvalues of H^(0).
8.5 Perturbation Theory Consists of a Set of Successive Corrections to an Unperturbed Problem
In this section, we shall derive the equation for a first-order correction to the energy.
In order to keep track of the order of our perturbation expansion, it is convenient to introduce a parameter λ into the Hamiltonian operator:
H^=H^(0)+λH^(1)
The factor λ is simply a bookkeeping device that will help us identify to what order our resultant perturbation equations are valid. We shall see that terms linear in λ give us what we call first-order corrections, terms in λ2 give us second-order corrections, and so on.
It’s a fact that ψn and En will depend upon λ. We assume that we can express the ψn and En as power series in λ, so that:
where O(λ3) means terms of order λ3 and higher. Notice that both terms in the first set of parentheses, the coefficient of λ0, are of zero order; all four terms in the second set of parentheses, the coefficient of λ1, are of first order; and so on.
Because λ is an arbitrary parameter, the coefficients of each power of λ must equal zero separately. The terms in the first set of parentheses equal zero. Let’s look at the coefficent of λ1:
H^(0)ψn(1)+H^(1)ψn(0)=En(0)ψn(1)+En(1)ψn(0)
The equation can be simplified considerably by multiplying both sides from the left by ψn(0)∗ and integrating over all space:
We can apply perturbation theory to a helium atom. For simplicity, we will consider only the ground-state energy. If we consider the interelectronic repulsion term, e2/4πϵ0r12, to be the perturbation, then the unperturbed wave functions and energies are the hydrogen-like quantities given by:
This integral is a little lengthy, the final result is:
E(1)=85Z(16π2ϵ02ℏ2mee4)
In units of mee4/16π2ϵ02ℏ2, we have:
E=E(0)+E(1)=−Z2+85Z
Letting Z=2 gives −2.750 compared with our simple variational result (−2.8477) and the experiment result of −2.9033. So we see that first-order perturbation theory gives a result that is about 5% in error.
It turns out that second-order perturbation theory gives −2.910 and that higher orders give −2.9037. Thus, we see that both the variational method and the perturbation theory are able to achieve very good results if carried far enough.
8.6 Selection Rules Are Derived from Time-Dependent Perturbation theory
The spectroscopic selection rules determine which transitions from one state to another are possible. The very nature of transitions implies a time-dependent phenomenon, so we must use the time-dependent Schrödinger equation:
H^Ψ=iℏ∂t∂Ψ
and ψn(r) satisfies the time-independent Schrödinger equation:
H^ψn(r)=Enψn(r)
Consider now a molecule interacting with electromagnetic radiation. The electromagnetic field may be written approximately as:
E=E0cos2πνt
If μ is the dipole moment of the molecule, then the Hamiltonian operator for the interaction of the electric field with the molecule is:
H^(1)=−μ⋅E=−μ⋅E0cos2πνt
So the H^ is:
H^=H^(0)+H^(1)=H^(0)−μ⋅E0cos2πνt
We will se below that the time-dependent term H^(1) can cause transitions from one stationary state to another.
To solve the equation, we will treat the time-dependent term H^(1) as a small perturbation. The procedure we will use is called time-dependent perturbation theory.
For simplicity of notation we will consider only a two-state system.
Ψ1(t)=ψ1e−iE1t/ℏandΨ2(t)=ψ2e−iE2t/ℏ
Assume now that initially the system is in state 1. We let the perturbation begin at t=0 and assume that Ψ(t) is a linear combination of Ψ1(t) and Ψ2(t) with coefficients that depend upon time:
Ψ(t)=a1(t)Ψ1(t)+a2(t)Ψ2(t)
where a1(t) and a2(t) are to be determined.
We substitute the equation into the time-dependent Schrödinger equataion:
Because H^(1) is considered a small perturbation, there are not enough transitions out of state 1 to cause a1 and a2 to differ appreciably from their initial values. Thus, as an approximation, we may replace a1(t) and a2(t) by
their initial values to get:
iℏdtda2=exp[ℏ−i(E1−E2)t]∫ψ2∗H^(1)ψ1dτ
( 微扰法的“微”字在此体现,由于扰动非常小,所以可以认为线性组合系数近似保持不变。 )
For convenience only, we will take the electric field to be in the z direction, in which case we can write:
Because we have taken E2>E1, the so-called resonance denominators cause the second term in this equation to become much larger than the first term and to be of major importance in determining a2(t) when:
E2−E1≈ℏω=hν
Thus, we obtain in a natural way the Bohr frequency condition we have used repeatedly. When a system makes a transition from one state to another, it absorbs (or emits) a proton whose energy is equal to the difference in the energies of the two states.
The probability of absorption or the intensity of absorption is proportional to the probability of observing the molecules to be in state 2, which is given by a2∗(t)a2(t).
But that is not applicable under normal conditions because the irradiating source consists of at least a narrow band of frequencies, so the equation must be averaged over this band.
When ω=ω12=(E2−E1)/ℏ, the P1→2(t) reaches its peak. So if g(ω) does not vary too strongly around ω12, then to a good approximation, we may take g(ω12) out from under the integral sign and write it as:
The spectroscopic absorption coefficient is the rate at which transitions occur, and so equals the time derivative of P1→2, or:
W1→2=2π[ℏ(μz)12E0z]2g(ω12)
This formula simply says that there must be radiation at the frequency ω12=(E2−E1)/ℏ for a transition to occur, which is just a formal statement of the Bohr frequency condition. The equation is a form of what is called Fermi’s golden rule.