《有机合成-切断法》章末总结3
第三篇总结,本篇主要总结一下各种大小环的合成。
1 饱和杂环
对于各种饱和杂环的合成,主要是通过分子内的亲核取代反应来实现,由于分子内反应容易发生,所以合适大小的环化是很有利的。
对于不同大小的环而言,五元环最易形成,六元环和三元环次之,七元环就很慢了,四元环更慢,可以参考下图来建立直观的感觉。
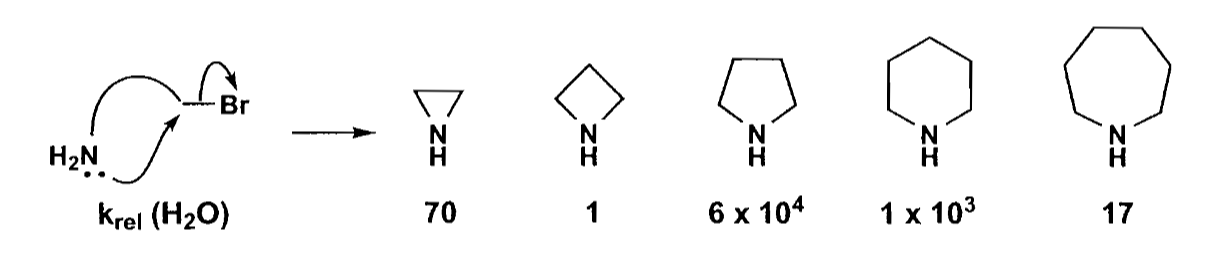
三元、四元等小环由于环张力的影响,在热力学上是很不利的,但是三元环由于官能团距离近,因此在动力学上又有优势;五元、六元环的环张力较小,成环后构型也稳定,热力学有优势,因此易于合成;七元环和更大的环虽然环张力小,热力学优势也不大,环化趋向一般。

下面列一下各种环的合成。
三元杂环主要是环氧化合物和氮杂环丙烷。
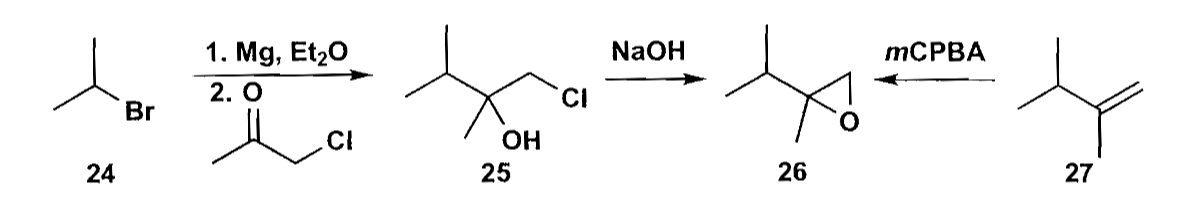

四元环由于其很大的位阻一般很难合成,不过在分子内无法成其他环的时候也可以成四元环,如下例。
五元环是最易形成的,来看两个例子。
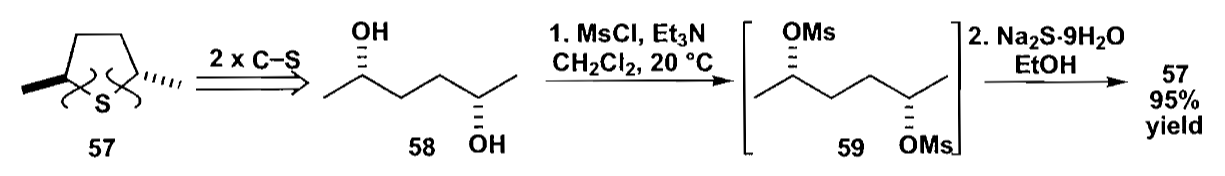
六元环的合成和五元环类似,同样是看两个例子。
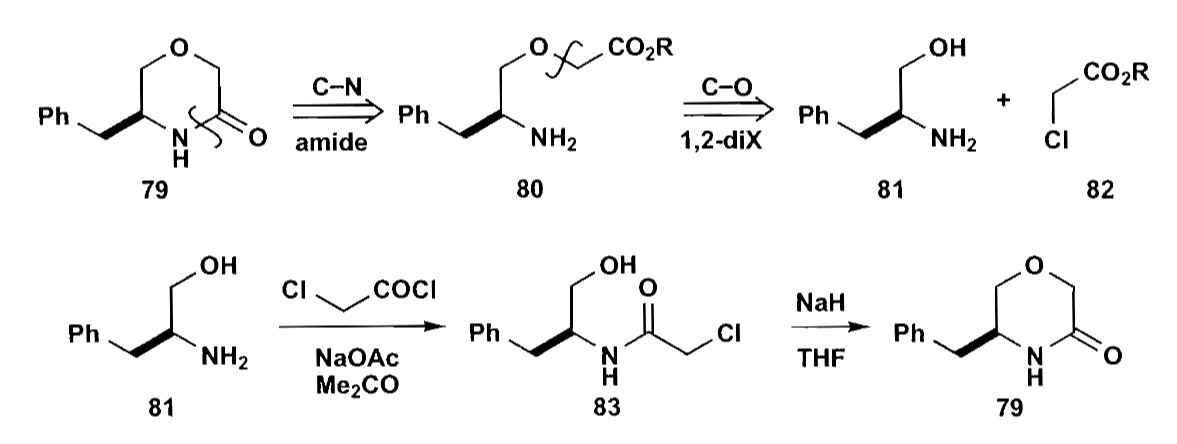
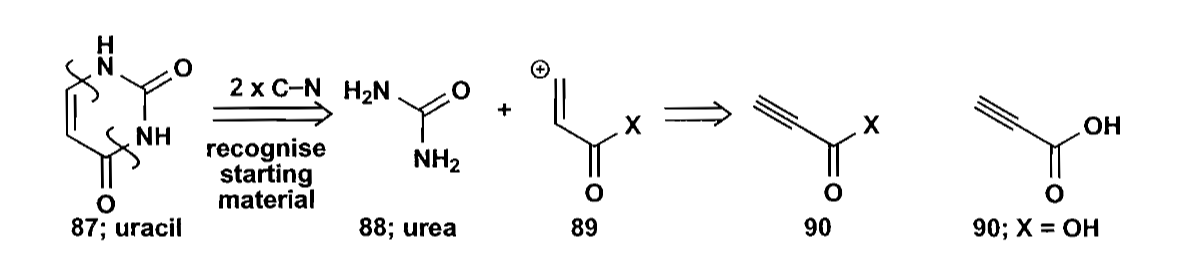
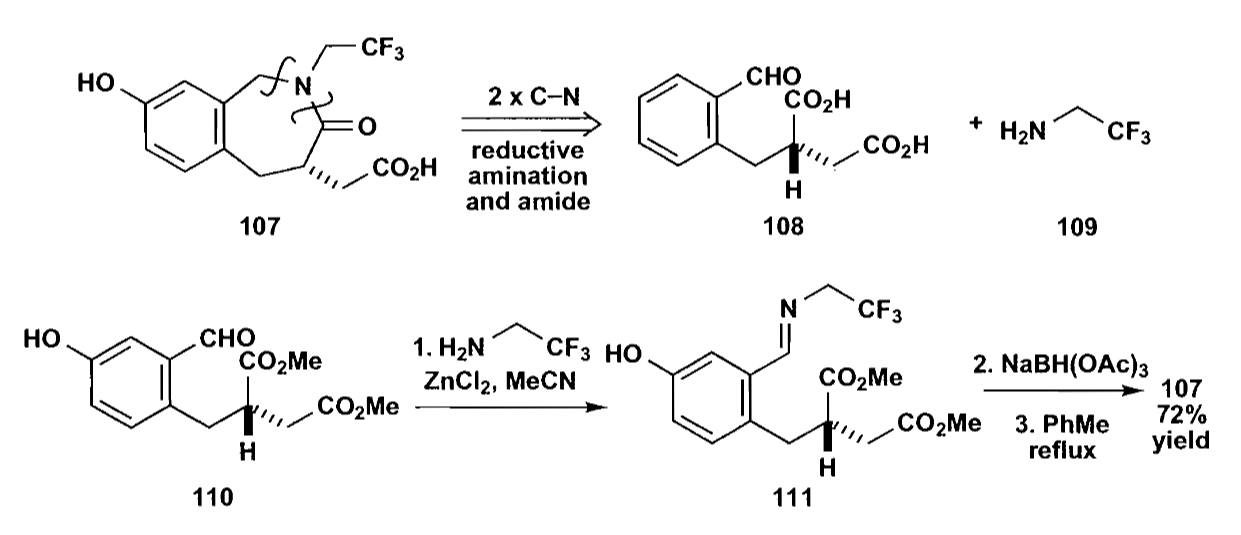
七元环,特别是苯丙氮杂七元环类,在药物合成中有重要作用。虽然七元环不太容易合成,但是仍要比分子间反应要好。同时,下面的例子说明,七元环的合成要比八元环容易一些。
2 三元环
从本节开始,后面讨论内容均为碳环的合成。首先是三元环。
三元环在动力学是有利的,在热力学上不利,因此成环的反应通常也能开环,所以可逆反应不适合用于合成三元环,但是烯醇α-碳的烷基化是不可逆的,因此这个反应可以用来合成三元环。
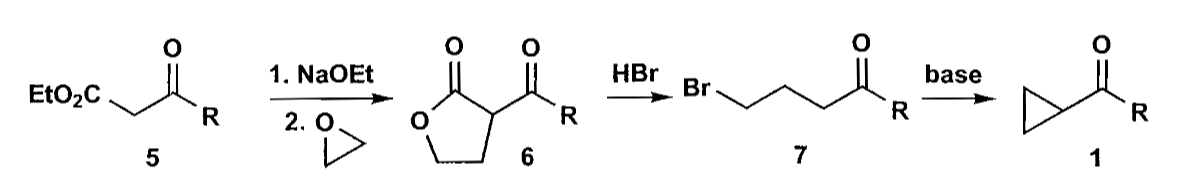
如上图反应中,二羰基化合物发生α-烷基化,并迅速形成五元内酯结构,再用酸水解,开环同时脱羧,得到化合物7,最后再用碱处理关三元环。产率可以达到82%。
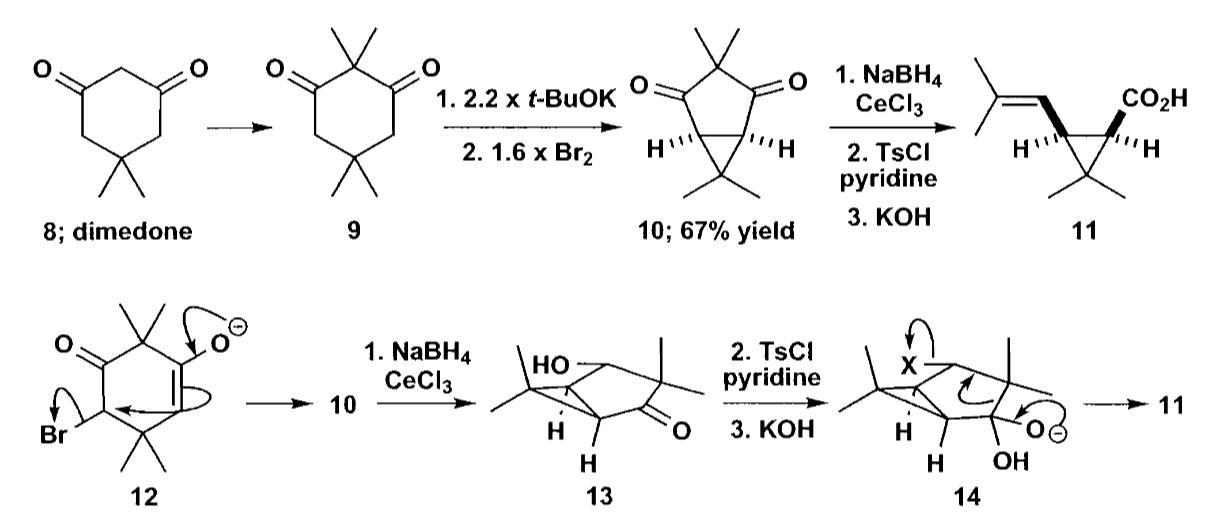
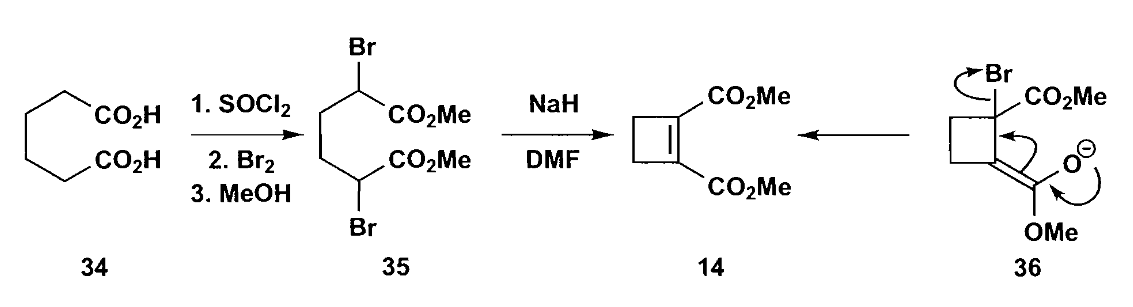
另一个例子是类似Favorskii重排(重排反应将在本章最后讨论)的成环方式,可以构建并环化合物,最终得到三元环。
合成三元环的方法除了简单的切断外,还有卡宾插入法。卡宾的制备有重氮法,α-消除法等,卡宾的制备在基础有机中介绍过,在此不详细介绍。至于插入反应,其实也没有什么好说的,还是看例子吧。
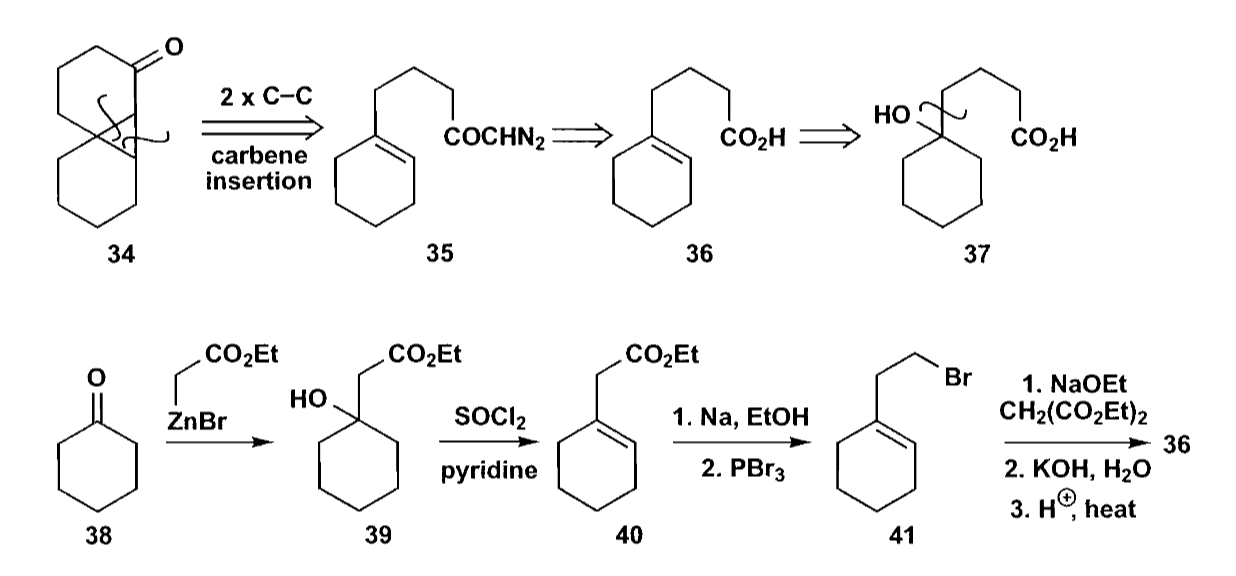
如上图反应,通过Reformastsky反应得到化合物39后,转化为化合物36,然后通过合成重氮化物,最终卡宾插入的反应来合成并环化合物34。
有些金属能够形成金属卡宾,同样也可以发生双键插入形成三元环。
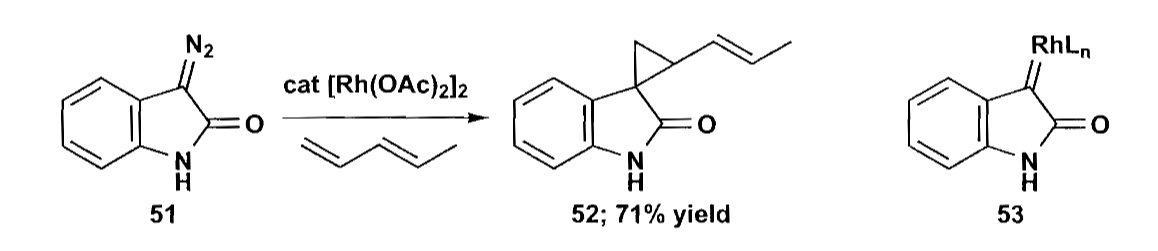
除了典型卡宾外,类卡宾试剂也是常用的选择。比如Simmons-Smith反应,在烯丙醇上反应时,会生成顺式三元环,这说明醇引导金属锌类卡宾插入烯烃。
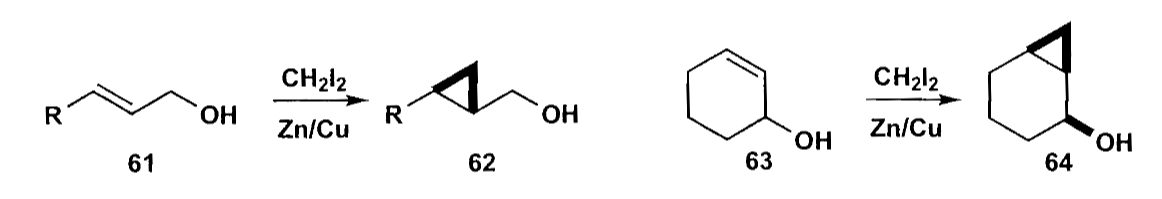
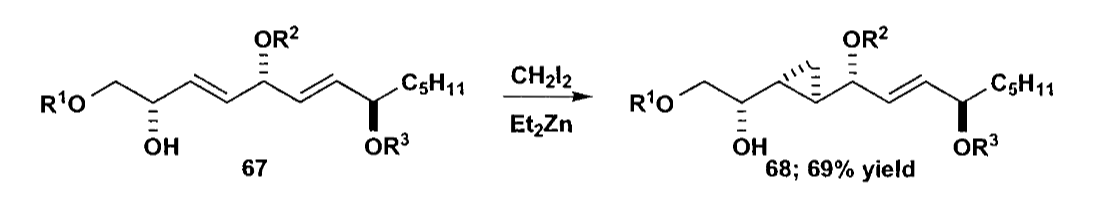
Takemoto合成的halicholactone就是一个带立体化学的例子,虽然反应物中有两个烯烃(下图化合物67),但是由于烯丙醇的协同参与,仅仅生成一个环丙烷。
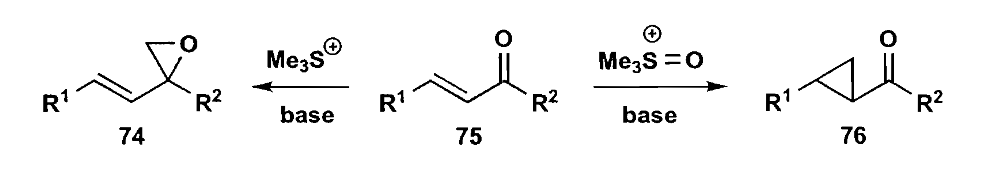
硫ylide也是构建三元环的好方式,通过硫ylide对不饱和羰基化合物的进攻,就能形成β-ylide取代物,进而通过烯醇烷基化把三元环构建完成。本质上还是烯醇α-烷基化的思路。
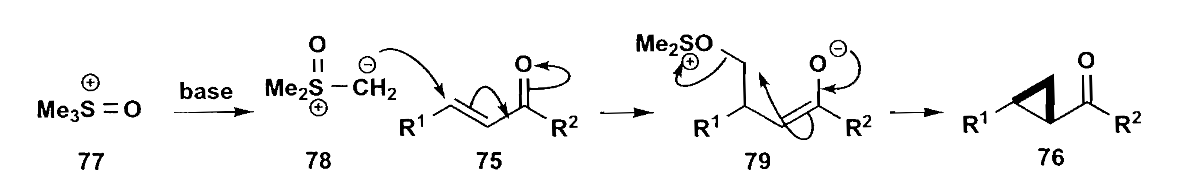
这里需要考虑一个问题,虽然硫化物是比较软的碱,但是也有可能发生直接加成羰基的副反应,最后会生成含氧三元环。为了得到环丙烷化合物,我们需要使用亚砜ylide。
一个更复杂的例子是Wills报道的halicholactone的合成,与上文Takemoto使用的方法不同,他使用了亚砜ylide来构建三元环。
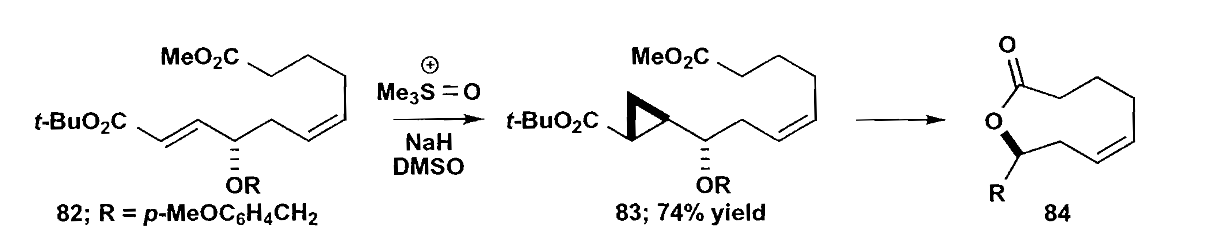
上面的反应没有体现出Simmons-Smith反应中导向的作用,因为羟基被烷基取代了。另外,化合物84中R基为含三元环的侧链。
上面提到的三元环的合成总体上可以分为两类,一是利用烯醇α-烷基化,其中离去基团的选择不同;二是卡宾的双键插入,其中卡宾的种类和生成不同。
3 四元环
四元环通过常规方法不易生成,所以一种可行的途径是光化学环加成法。常规的D-A反应大家都已经非常熟悉,[4+2]的环加成生成六元环结构,要想生成四元环,则需要进行[2+2]的环加成,由于轨道对称性的要求,4π电子的环加成反应需要在光条件下进行。
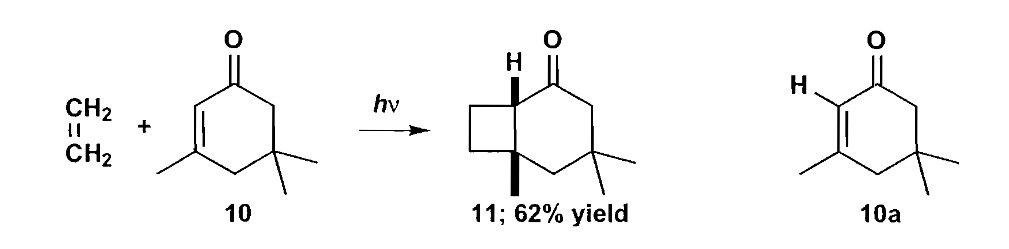
对于环丁烷类化合物的合成,我们需要考虑如何切断的问题,不同切断方式的合成难度不同,选择合理的切断往往能让合成事半功倍。
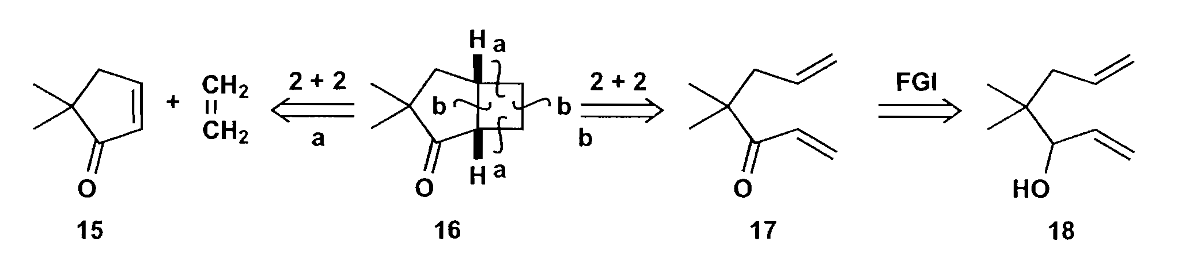
既然提到了环加成,那么区域选择性和立体选择性是绕不开的话题,[2+2]环加成的区域选择性可以使用和[4+2]环加成相同的方式来作判断,注意[2+2]中有激发态参与反应,因此激发态物质的极性反转,可参考下图反应,
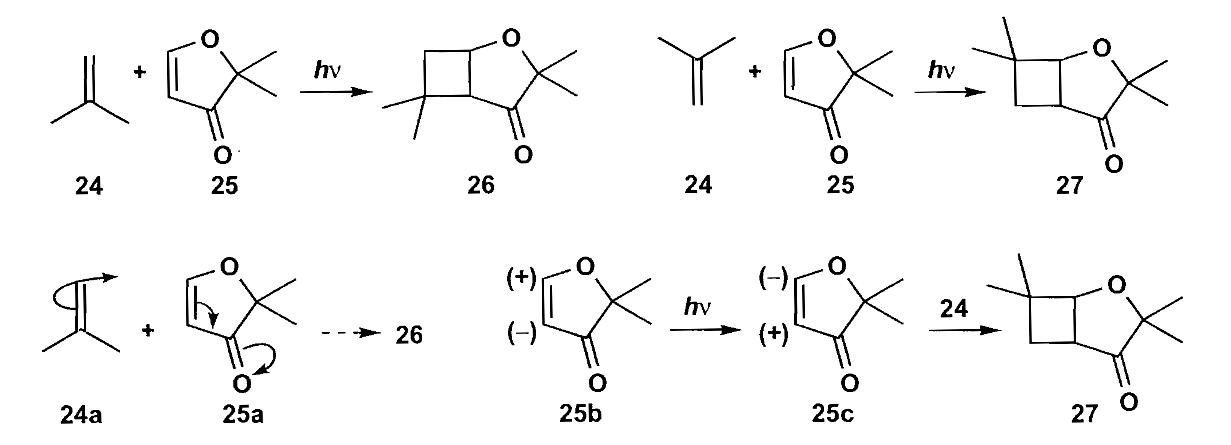
在上图所示反应中,化合物27是唯一的反应产物,没有化合物26的生成,下面的极性表示了反应的区域选择性。
分子内加成同样满足反应的区域选择性,这可能会导致产物具有一个扭曲的构型,例如Solanoeclepin A的合成中间体(即下图中化合物32)的合成。
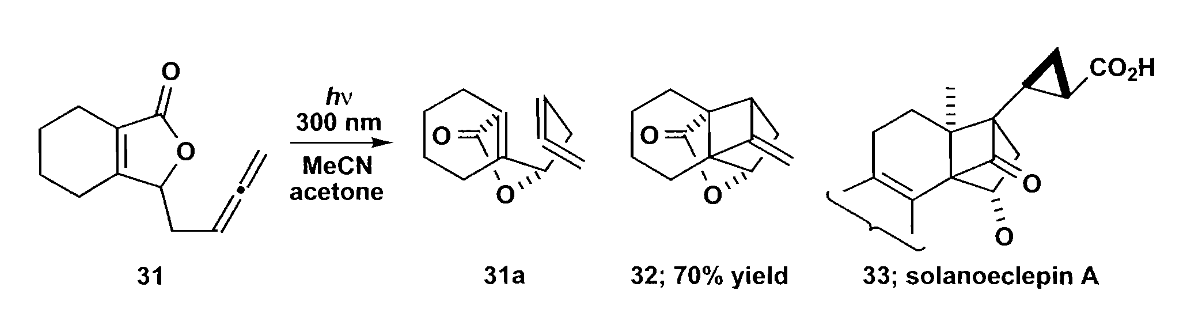
当然,如果扭曲张力实在太大,分子也有可能会选择不符合区域选择性,但是张力更小的方式成环,比如下面的例子。
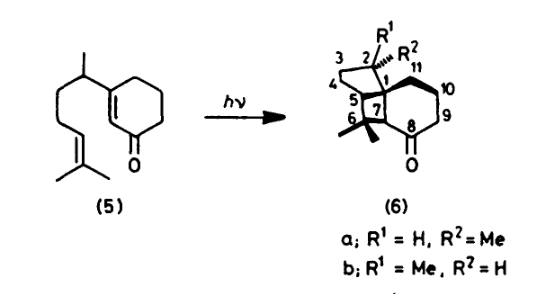
( 上图来自 J. Chem. Soc., Perkin Trans. 1, 1979, 1407-1410 ,之所以没有用书上的图是因为书上画的反应图有问题,反应物少一个碳… )
离子型反应生成四元环也不是完全不行,但是不如环加成反应通用。
除了光反应的环加成,还有热反应环加成可供选择,但是不同于普通烯烃,热反应环加成需要使用烯酮才能进行。对于一般烯烃来说,热条件的[2+2]环加成的同面-异面加成是允许的,但是位阻太大难以发生,烯酮正交的π轨道允许发生同面-异面加成,所以可以直接在热条件下发生。烯酮的活性很高,因此通常不会被分离出来。
二氯乙烯酮和顺/反式环己烯的反应表明这是一个协同反应。
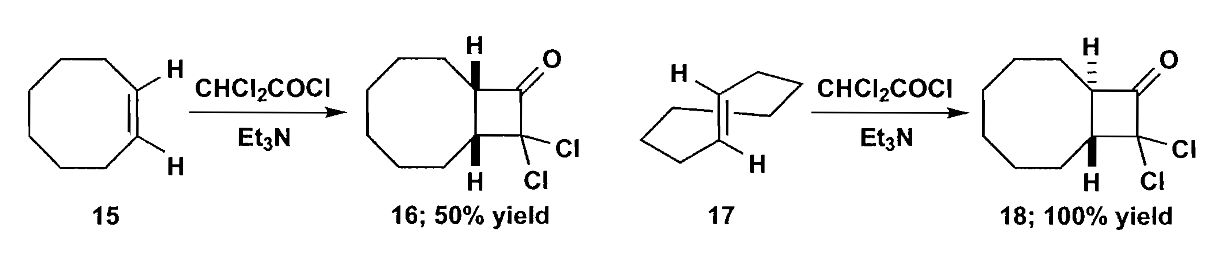
4 五元环
由于五元环在动力学和热力学方面相对开链化合物都有优势,所以五元环的羰基缩合是比较容易的,我们首先讨论通过羰基缩合构建五元环的方法。
环戊烯酮可以被切断成1,4-二羰基化合物,但是需要注意成环时的区域选择性问题。
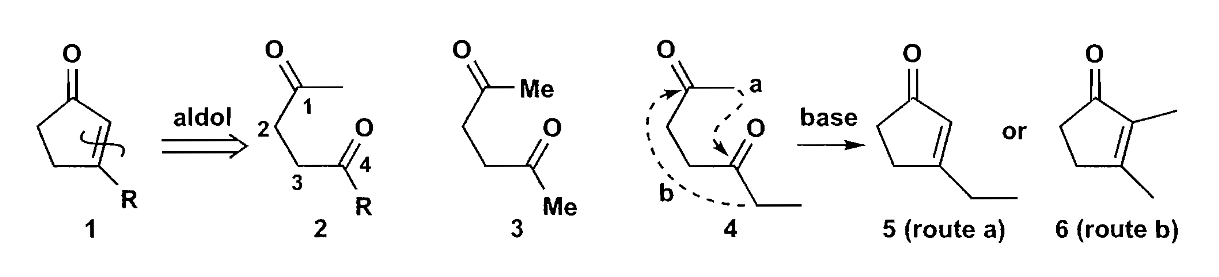
上图中可能会生成不同的五元环不饱和酮,化合物6是多取代的热力学烯烃,但是优势并不大。
分子间反应也是能发生的,如下图反应所示。
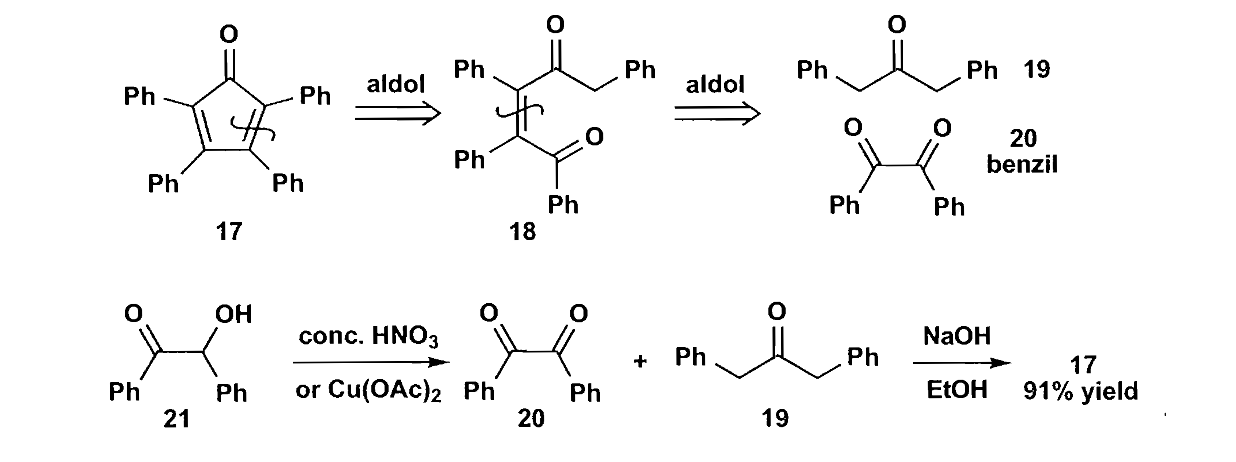
1,6-二羰基化合物也可用于五元环的合成,最经典的例子就是环己烯氧化切断双键生成二羧酸结构,酯化后进行分子内酯缩合得到缩环产物。
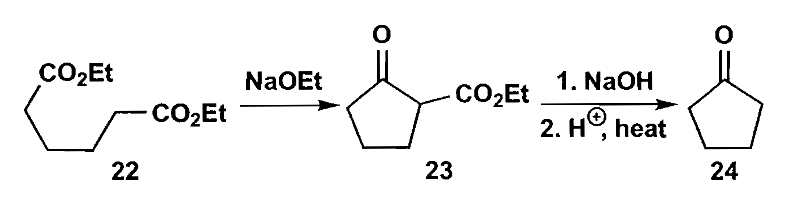
下面讨论一个稍复杂的例子,对于下图化合物27,可以转化为化合物29,其为一种天然产物苎烯。但是此处有两个问题需要考虑,怎样选择性切开一个双键而另一个不受影响以及怎么控制环化。
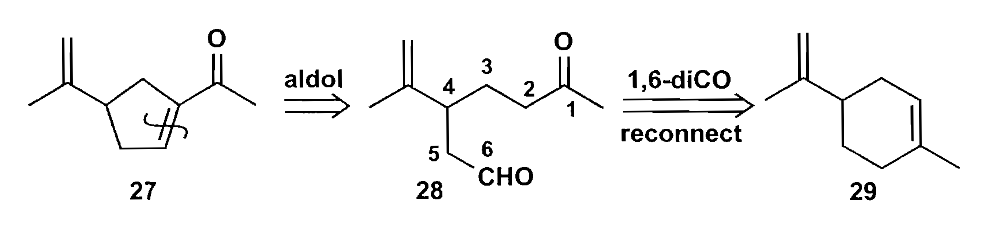
第一个问题的答案是选用具有选择性的反应,例如环氧化,环氧化会优先发生在环中取代基更多的烯烃上,这正是我们想要的结果,进而就可以切断生成1,6-二羰基化合物。
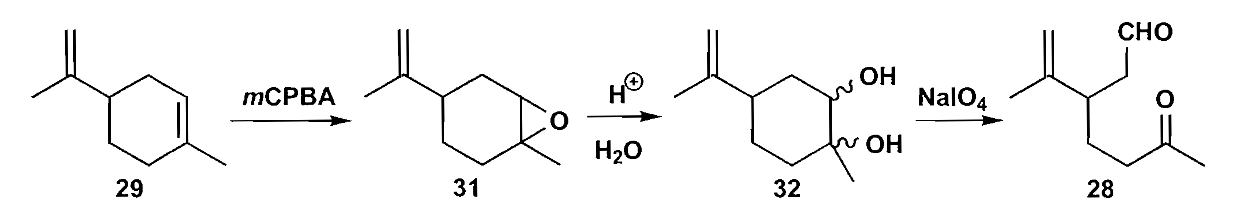
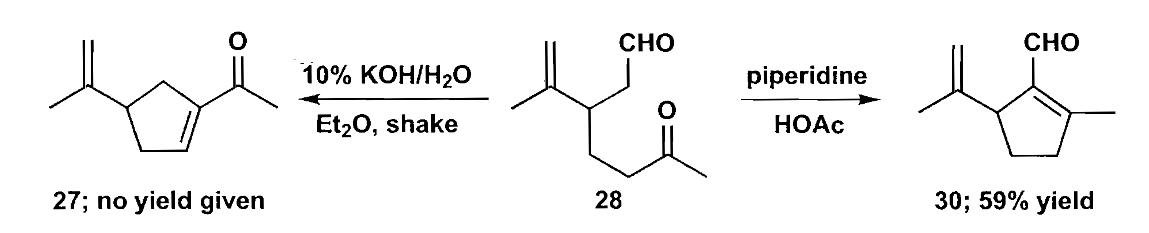
化合物28的关环同样需要选择性调控,一般而言,使用氢氧化钾这样的较强的质子碱条件下,反应是可逆的,生成热力学控制的更稳定的酮27;而在弱胺碱和弱酸组成的缓冲体系中,只有高活性的醛能够烯醇化,因此得到动力学控制产物30。
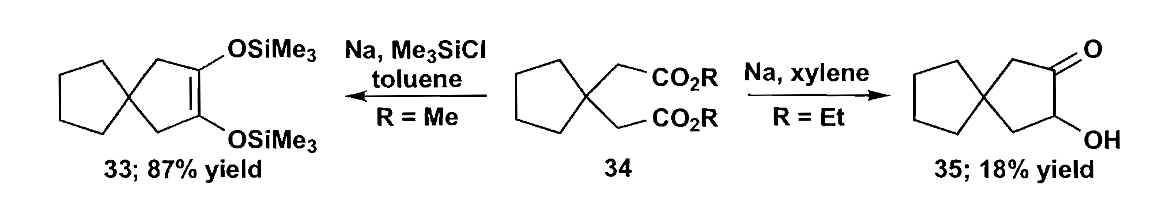
1,5-二羰基化合物也可以通过偶联来合成五元环,即酮醇缩合反应,在硅醚辅助下可以以很高产率生成五元环。
当然,还有最直接的连续共轭加成合成环戊烷,通过合理的串联加成反应构建五元环结构,如下图所示反应最终得到81%收率。
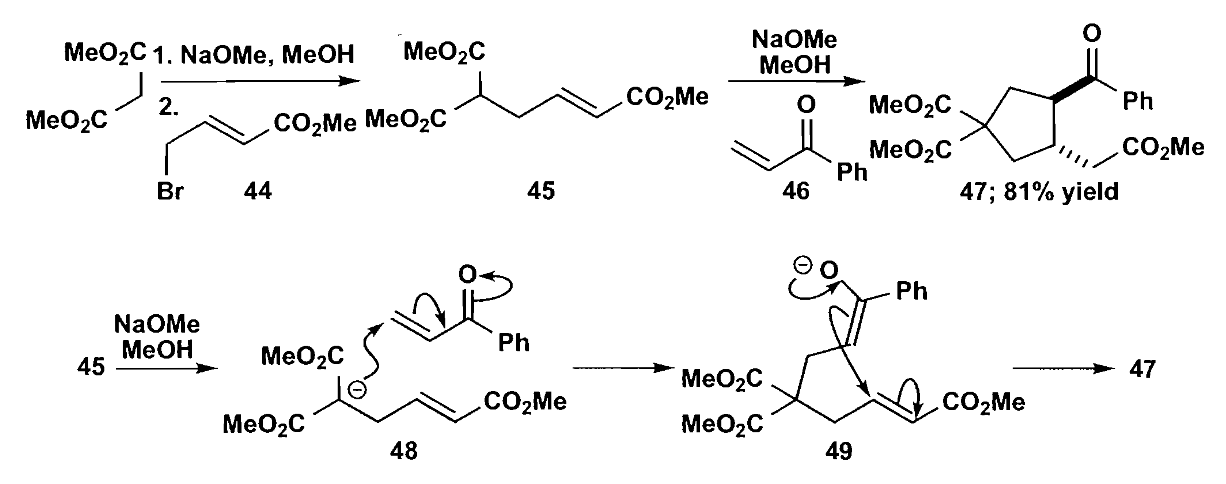
上文介绍了通过羰基化合物缩合构建五元环的反应,下面再讨论一些不同的方法,比如周环反应。
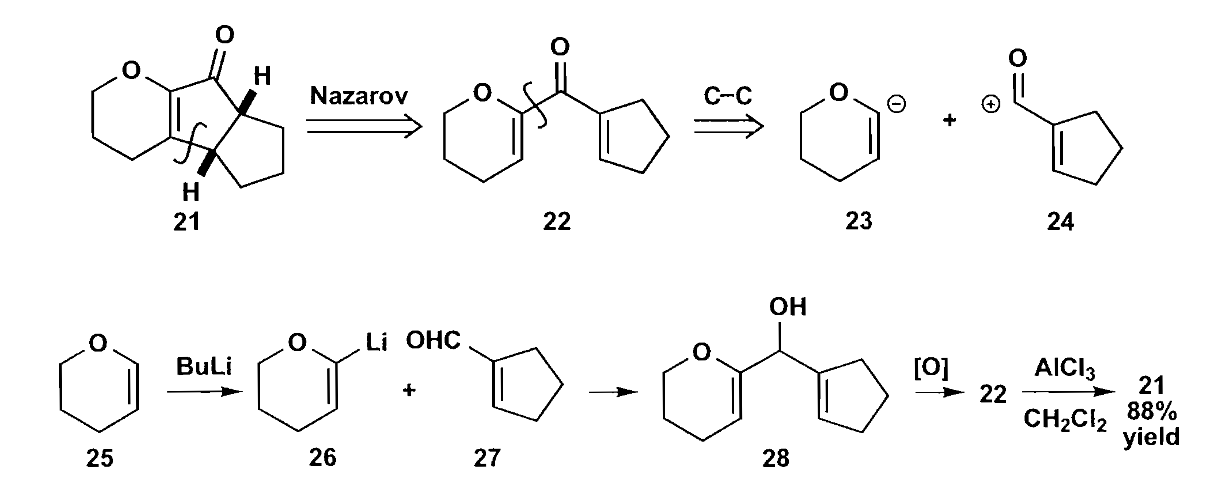
Nazarov环化是借助电环化关环生成五元环的重要方法之一,戊二烯正离子类型的化合物在酸性条件下很容易环化为五元环,看一个合成例子。
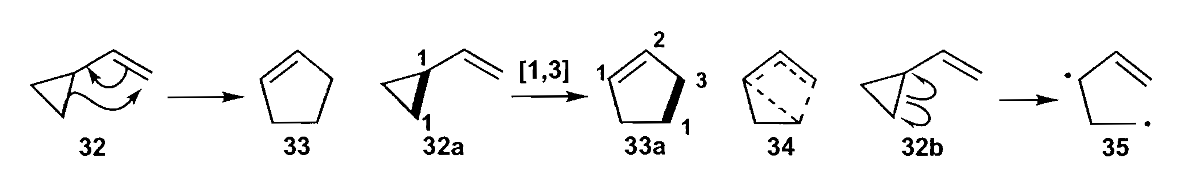
σ迁移也能用于合成五元环,乙烯基环丙烷结构发生[1,3]-σ重排即得到环戊烯,该反应条件需要加强热(一般>300℃)。该反应机理尚存争议,有人认为三元环中的碳碳单键均裂形成双自由基中间体。
这种切断并不常见,看一个用于合成的例子。
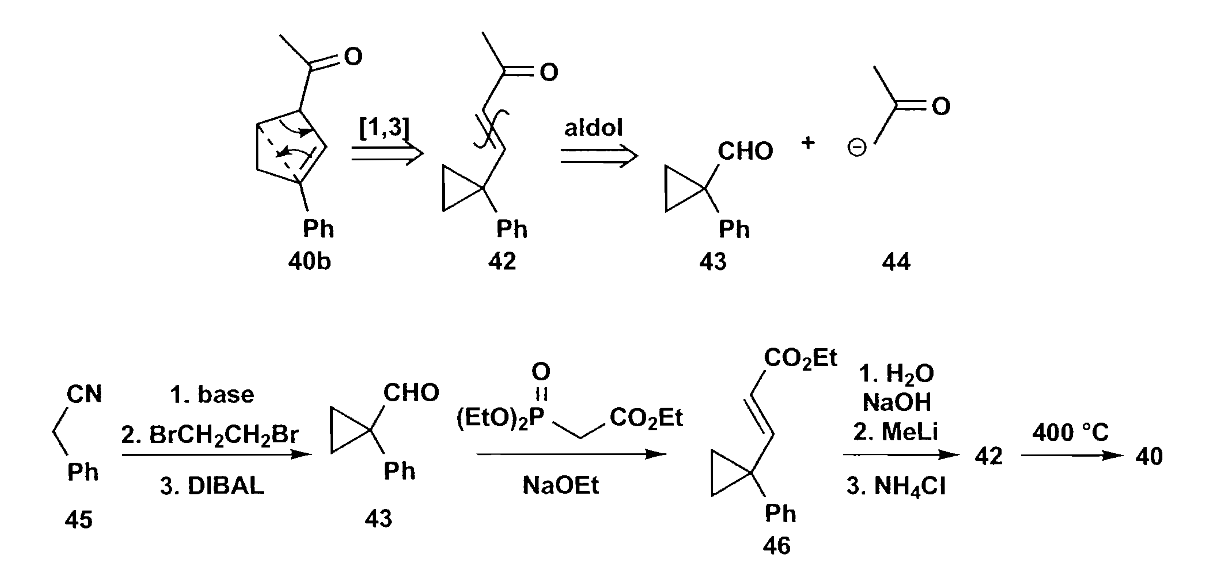
最后一步重排反应需要400℃高温,条件非常苛刻,因此在合成中用途较少。
5 六元环
构建六元环的方法介绍三种,首先是羰基化合物的缩合,典型反应是Robinson增环,有机催化剂的发展使得很多反应给出单一的对映异构体产物。
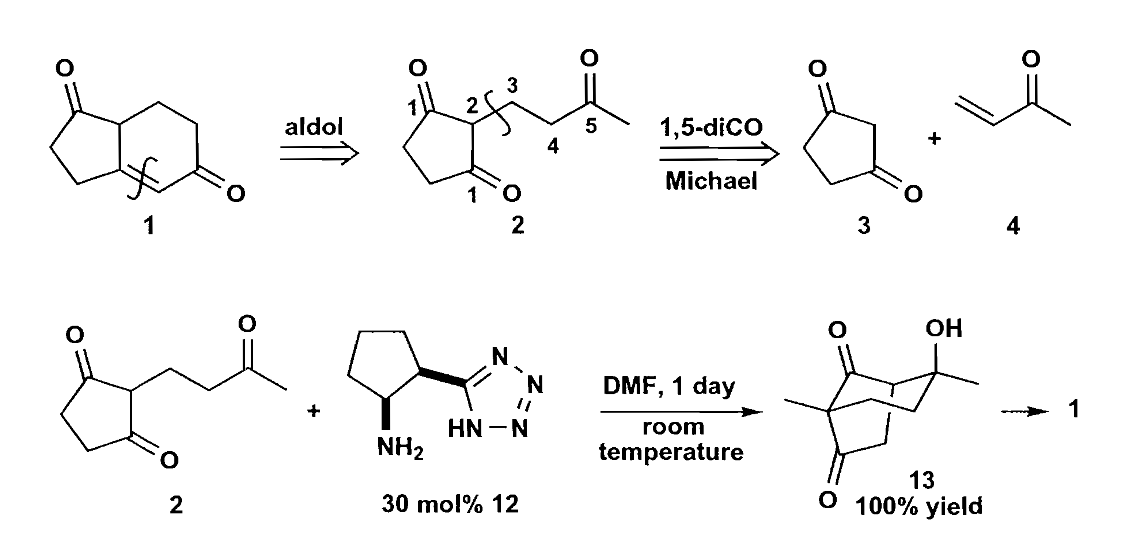
当然Robinson关环绝不是唯一的离子型反应,六元环的成环方法多种多样,比如上文介绍的Nazarov环化生成的碳正离子被捕获也可以发生自身环化,见下例。
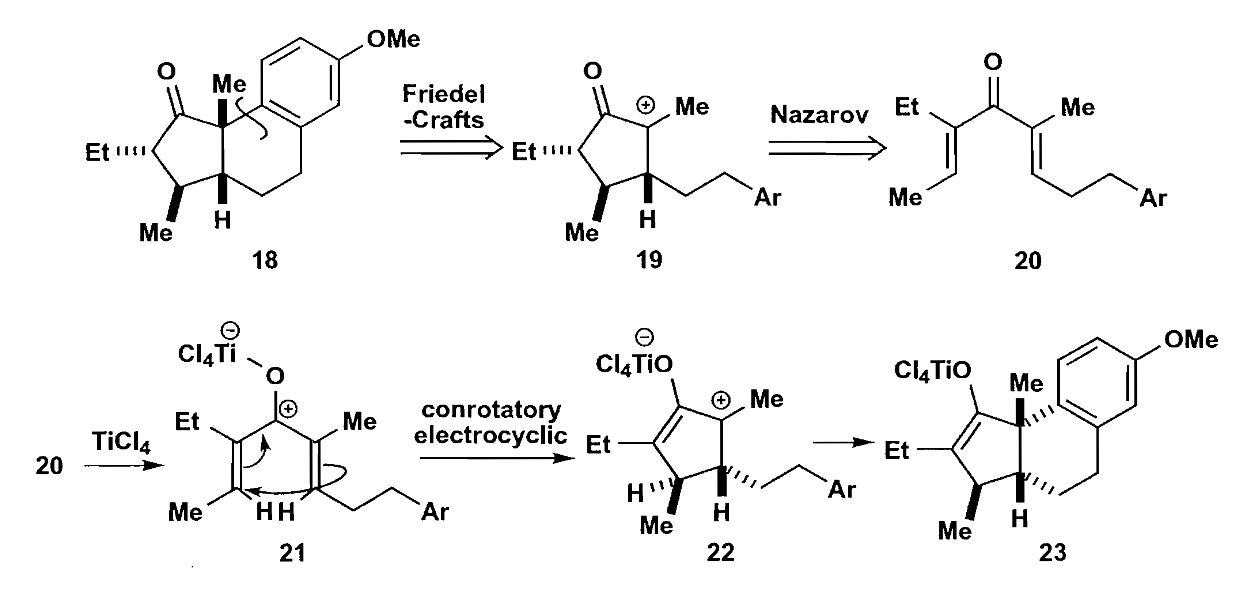
上图反应的切断并不常见,只有意识到这个Nazarov环化后才算合理,这个六元环的形成本质上利用了这个位置合适的碳正离子完成成环。
第二种成环思路是利用D-A反应,这个反应在各种成环合成中屡见不鲜,同时能很好地控制立体选择性,是非常好的方法。
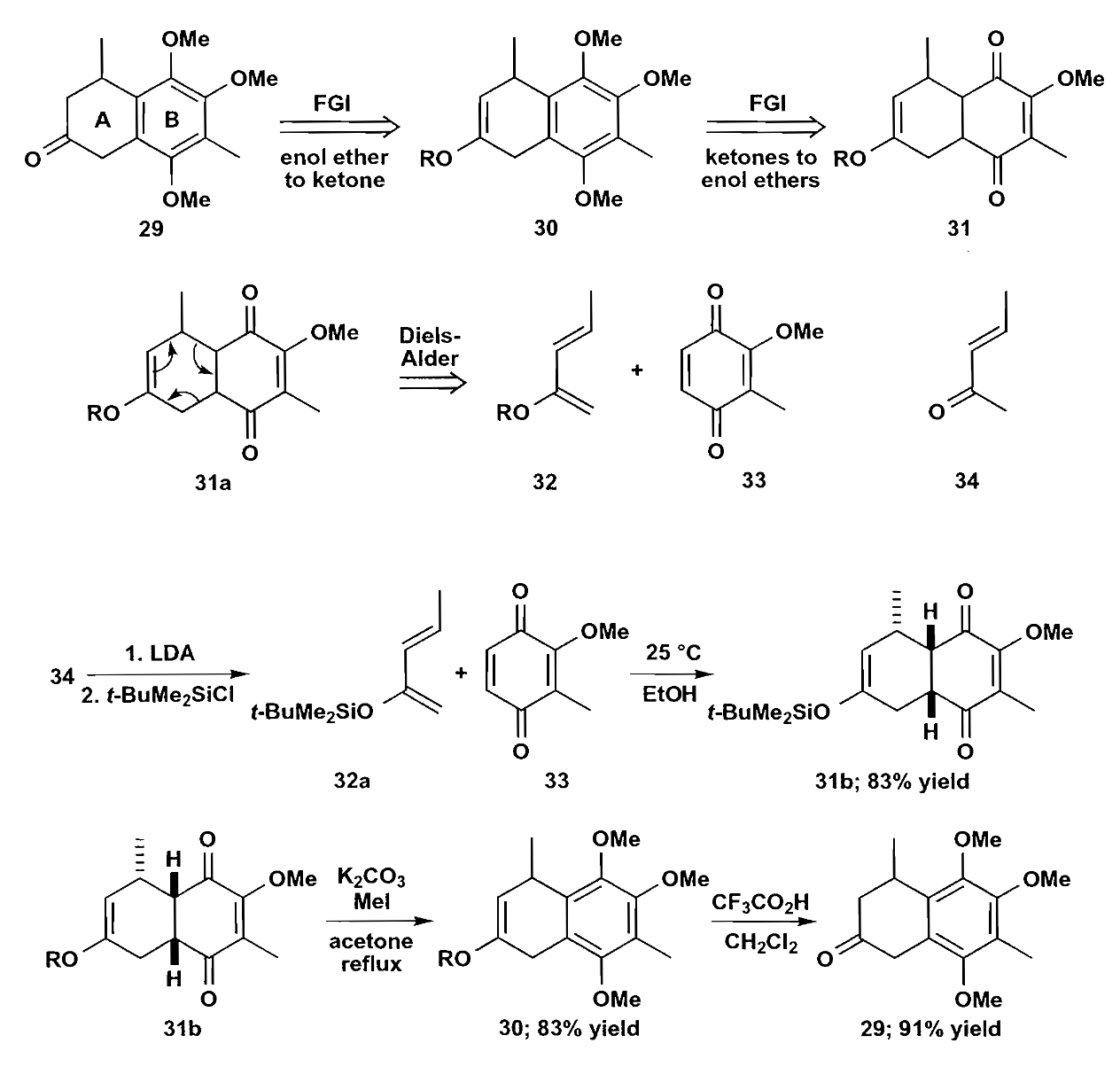
上图反应的区域选择性值得考虑,其“对位选择性”体现在甲氧基和上方羰基(即与甲氧基相连)之间。
再看看抗生素Guanacastepene骨架的合成,这个例子说明了D-A反应对于骨架的巨大影响,导致逆向切断有时候看上去不那么显然易见。

可见尽管上图中化合物36具有比较明显的环己烯结构,但是实际的合成方法并不是切断使用D-A,而是另外构造了新的环。
第三种思路是利用现有的环转化为目标六元环,例如芳环的还原,一般来说,要想破环芳香性不太容易,所以往往需要比较强的条件,比如金属催化加氢或者强还原剂等。
看两个例子:
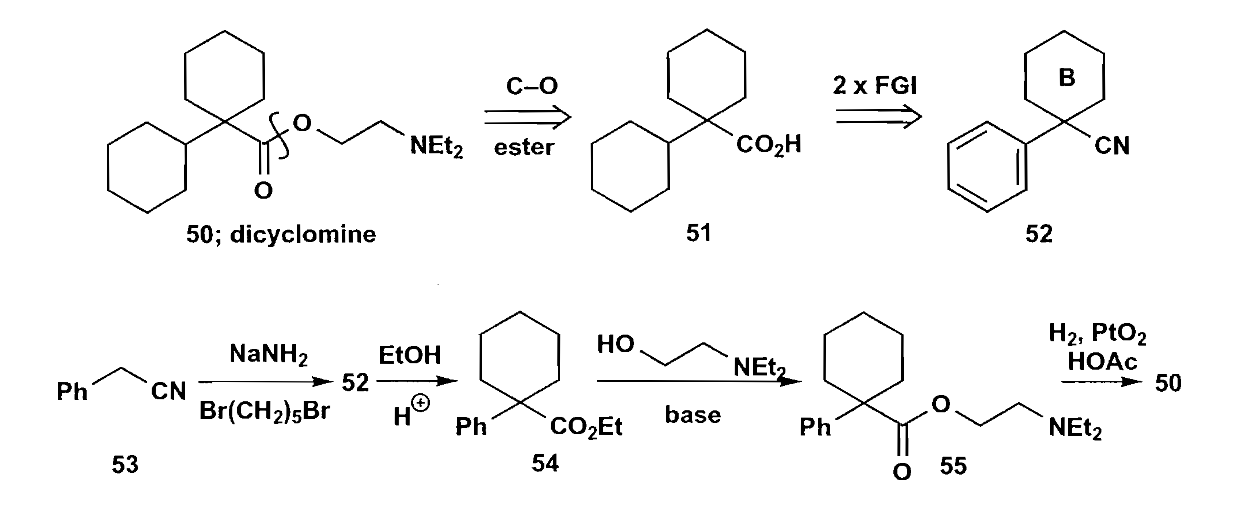
第一个例子利用了芳环的完全还原,对于化合物50来说,两个环中一个环是螺环,无法通过芳环还原形成,另一个环单独连在外面,常规方法不易合成,此时就很适合芳环完全还原法。
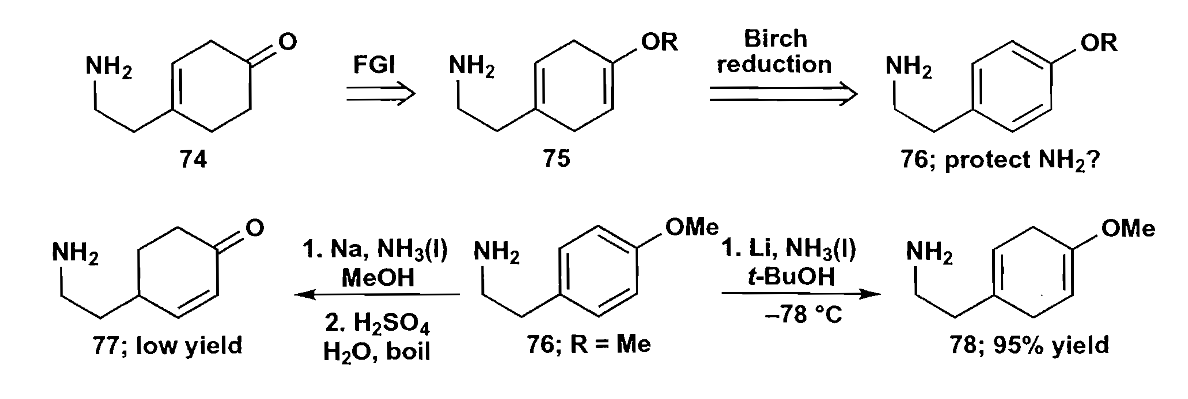
第二个例子是细胞毒素maritidine的前体的合成,这种取代环己烯的结构可以利用苯环的不完全还原来合成,典型反应如Birch还原。
6 环的重排
如果目标分子的碳骨架难以构建时,我们可以选择使用常规方法合成一个稍微不同的骨架,再利用重排反应合成所需的环,本节介绍几个环的重排反应 。
环的重排可以从环大小改变来分为扩环反应和缩环反应,当然有一些反应/试剂既能扩环也能缩环,我们会分开介绍。
首先是利用重氮烷烃的扩环反应,比较多见的例子是从易合成的六元环扩环到七元环。
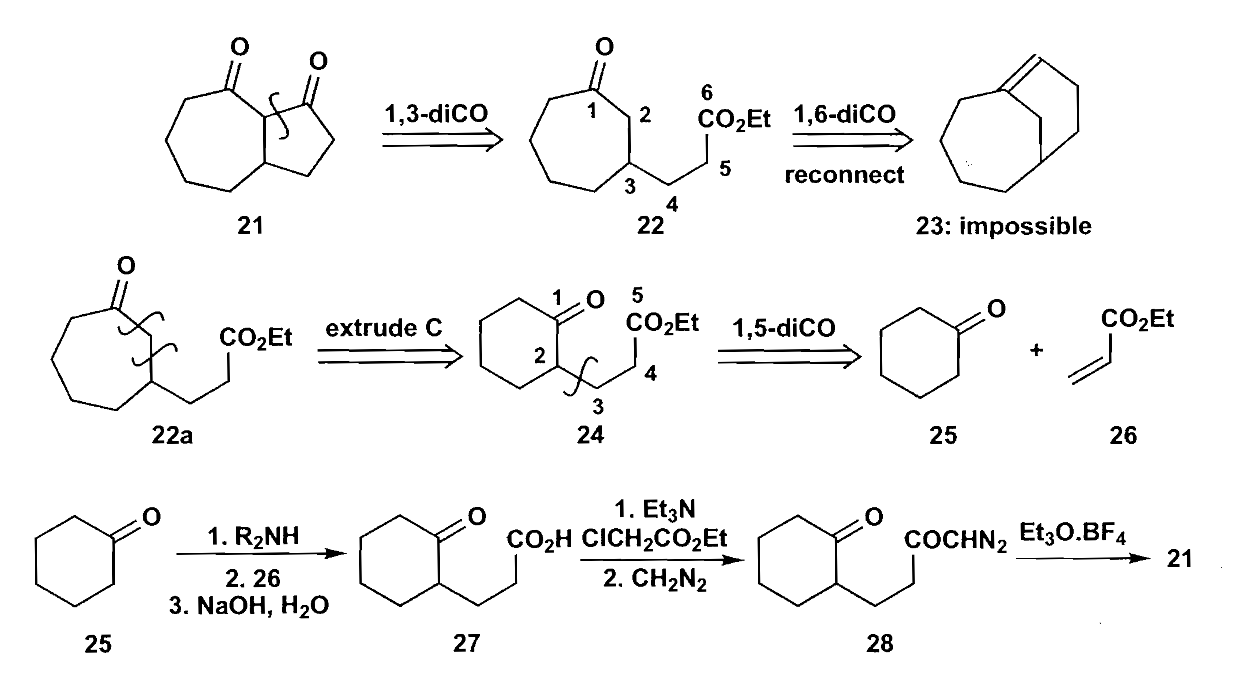
关键的扩环重排一步的机理是重氮甲烷的碳进攻羰基,形成一个含氧负离子和重氮离去基团的中间体,然后分子内转移得到产物。
pinacol重排也是相似的机理。
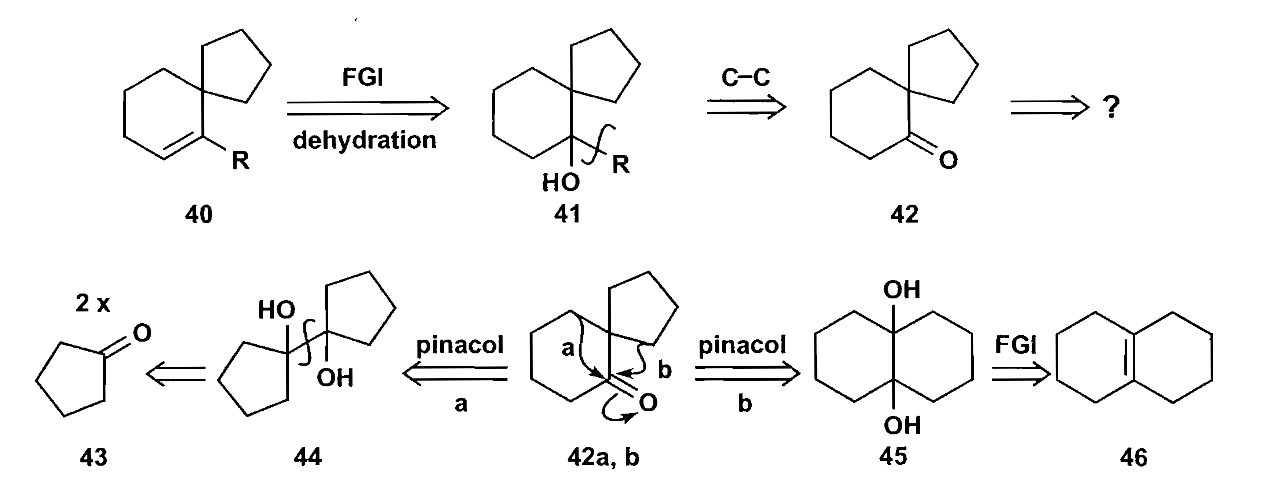
上图中合成的化合物40可以继续利用重排反应扩环得六元环螺环,使用的是Beckmann重排,与之类似的反应还有Baeyer-Villiger反应,这里只给出前者的例子。
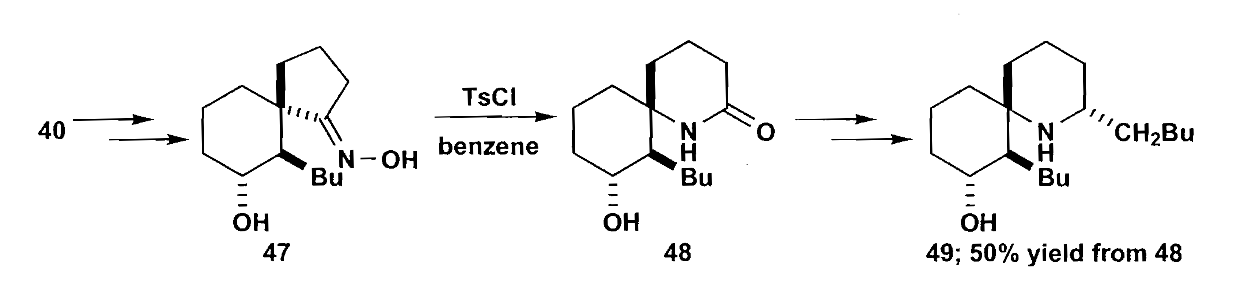
扩环反应说完了,接下来是缩环反应。
首先是环氧化合物的重排,环氧化合物在Lewis酸催化下开环生成碳正离子,这样的碳正离子可以被分子内迁移捕获,生成缩环的醛。
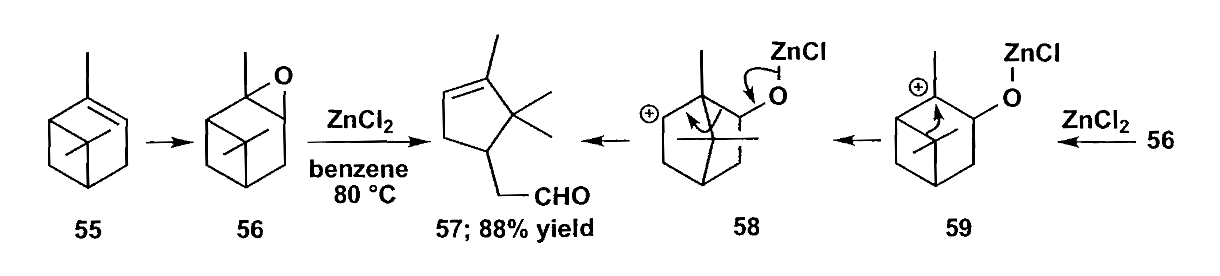
前文提到的重氮烷也能进行缩环,如下图所示的junionone的合成,难以合成的四元环可以通过五元环缩环实现,缩环时重氮基作为离去基团应该直接与环相连。
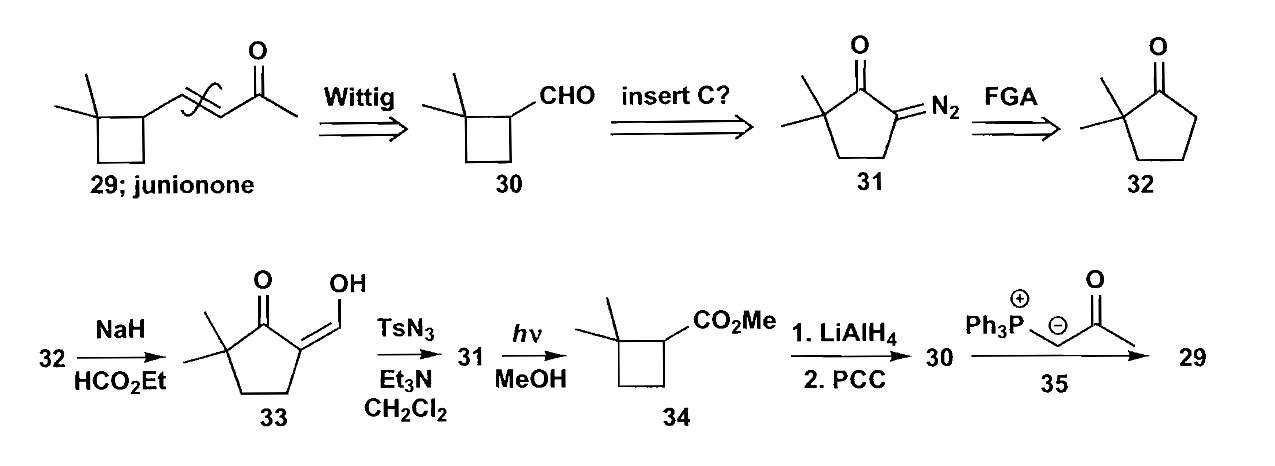
上图中从化合物33转化为化合物31的反应为Regitz重氮转移反应,这是常用的生成α-重氮酮的方法。
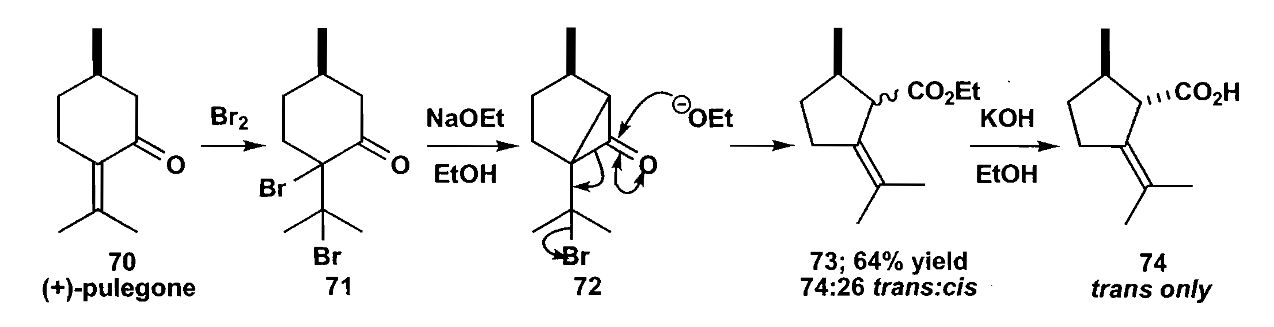
最后是Favorskii重排,与前两者不同,Favorskii反应是一个经由负离子中间体的缩环重排。
总结
环的合成通常是分子内反应,所以在动力学上往往有一定优势,因此常规大小的环是比较容易合成的,方式也比较多样。
对于小环或者大环就往往需要各种特殊的策略来合成,这些特殊的策略也会影响到路线的整体选择。尤其是小环的特殊合成方法往往决定合成策略。