《中级有机化学》章末总结6
第六章为亲核取代反应。拖了很久了。
第6章 亲核取代反应
6.1 饱和碳原子上的亲核取代反应
发生在卤代烷、醇、磺酸酯等有机化合物的饱和碳原子上亲核取代主要有两种机理:单分子亲核取代反应和双分子亲核取代反应。除此之外,也有分子内亲核取代反应。
反应的基本机理不必赘述。反应经历碳正离子中间体,碳正离子一旦形成就会涉及碳正离子的重排,形成更稳定的碳正离子。不过多数情况下,反应以构型翻转产物为主,这可以从离子对理论进行解释。
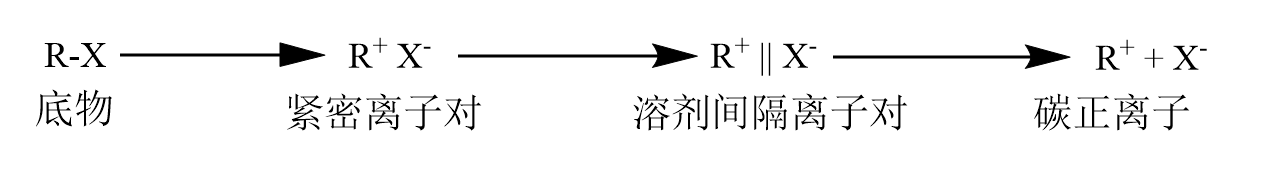
只有自由的碳正离子才会以均等的概率从两个方向接受亲核试剂的进攻,其他情况下,离去基团都倾向与从离去基团背面进攻,因此主要产物为构型翻转产物。
溶剂对反应的影响取决于反应的决速步是电荷集中的、还是电荷分散的过程。
反应是一步完成的协同反应,机理同样不必赘述。反应中经历一个五配位的过渡态,得到中心碳原子构型翻转的产物,这种构型翻转称为Walden翻转。
由于位阻的关系,叔卤代烷不发生反应,但也有例外:
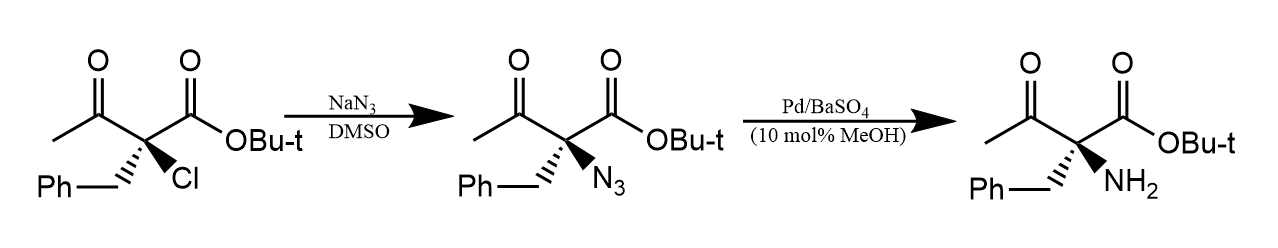
本反应能发生的原因有两点:一是因为底物中两个羰基的吸电子能力很强,键极化很强;二是因为叠氮根负离子的位阻很小。
当底物是烯丙基卤代烃、炔丙基卤代烃、苄基卤代烃时,发生反应的速率显著加快。这是因为亲核试剂和离去基团带有部分负电荷,可以通过和的相互作用而得到分散。因此,不饱和键的存在有效地降低了过渡态的势能,使反应更易发生,也就是我们常说的活化作用。
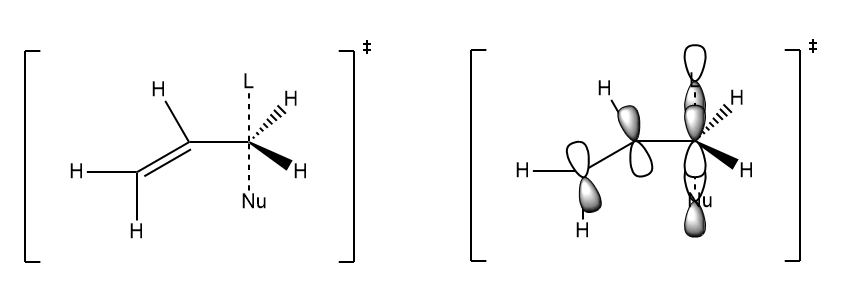
烯丙基卤代烃发生反应的同时,可以发生反应,发生烯丙基重排。
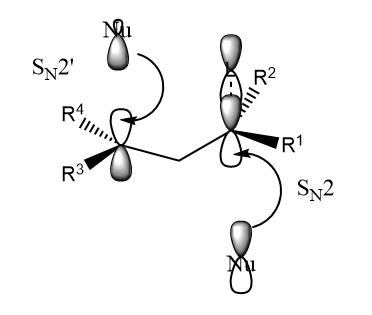
一般情况下,亲核试剂从离去基团的同侧进攻,因为烯丙基正离子的轨道的两端是相位相反的,进攻的时候,亲核试剂从离去基团的异侧进攻;进攻时,亲核试剂从离去基团的同侧进攻,才满足轨道方向性的原则。
和的竞争反应可以通过亲核试剂的结构进行控制,基团增大时,易发生反应。
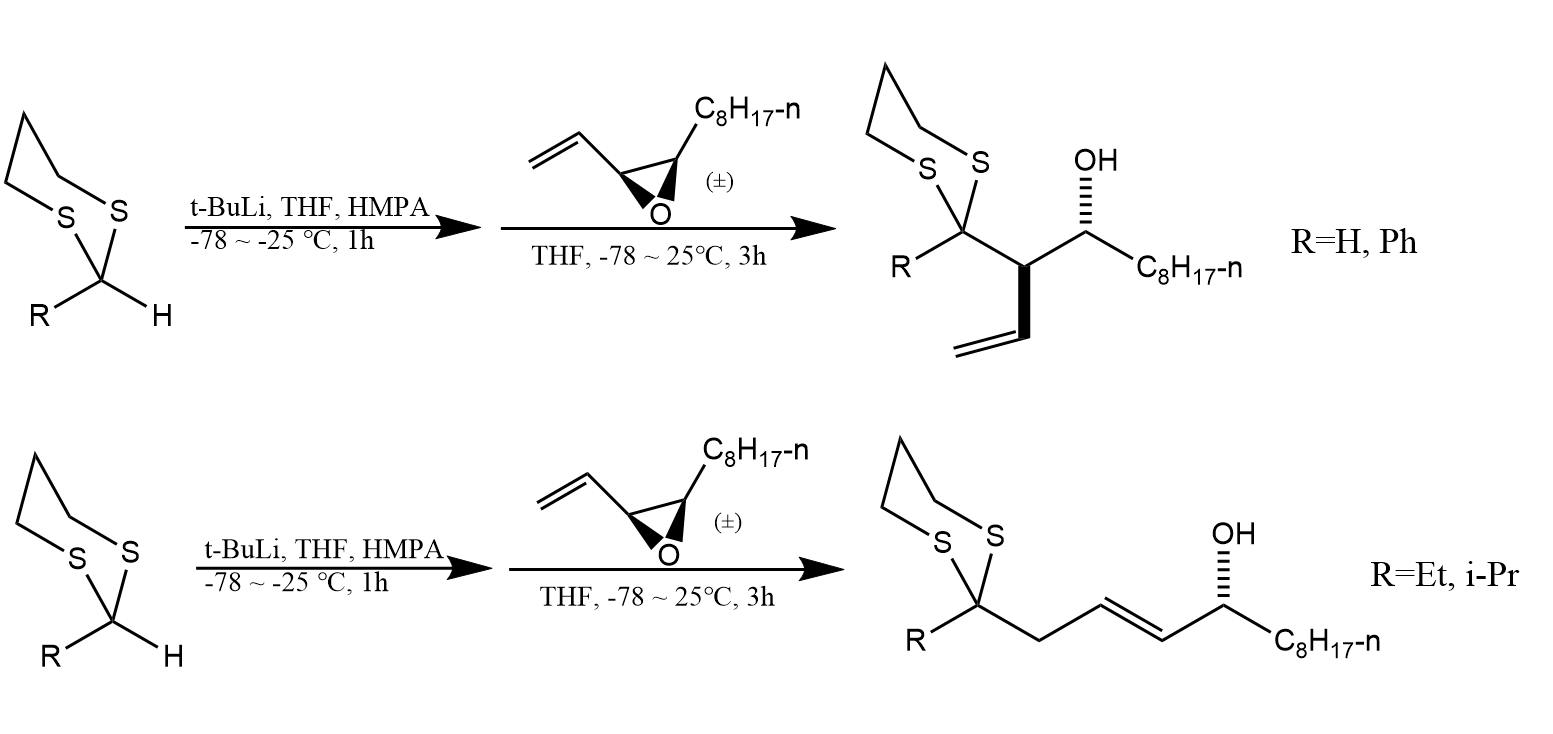
醇与二氯亚砜的反应是将醇转化为氯代物的常用方法。在二氧六环中,仲醇与二氯亚砜反应,经历两次后得到构型保持产物。若在吡啶中进行反应,则只发生一次反应,故得到构型翻转产物。
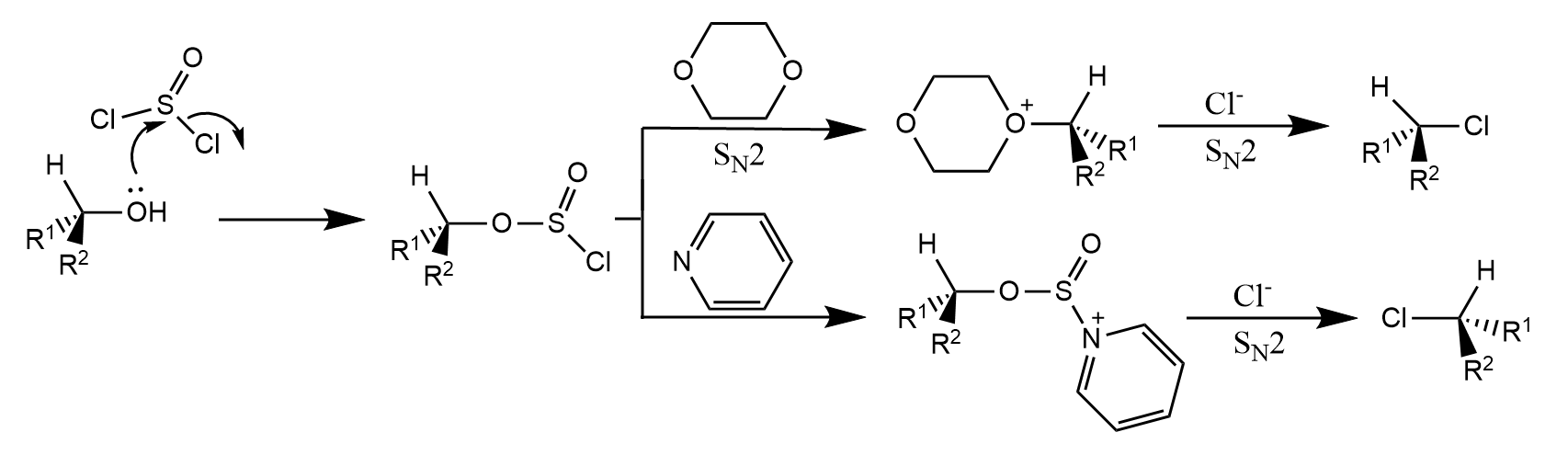
经典的Arbuzov反应和Gabriel反应是两个重要的例子:
亚磷酸酯和卤代烃在加热条件下生成膦酸酯的反应称为Arbuzov反应。
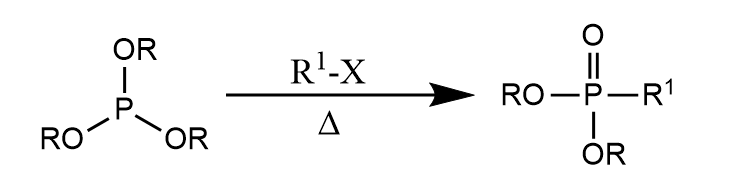
邻苯二甲酰亚胺的钾盐在非质子性溶剂中和空间位阻小的伯卤代烃或仲卤代烃发生亲核取代反应得到N-烷基化的邻苯二甲酰亚胺,通过酸/碱/肼处理得伯胺称为Gabriel反应。
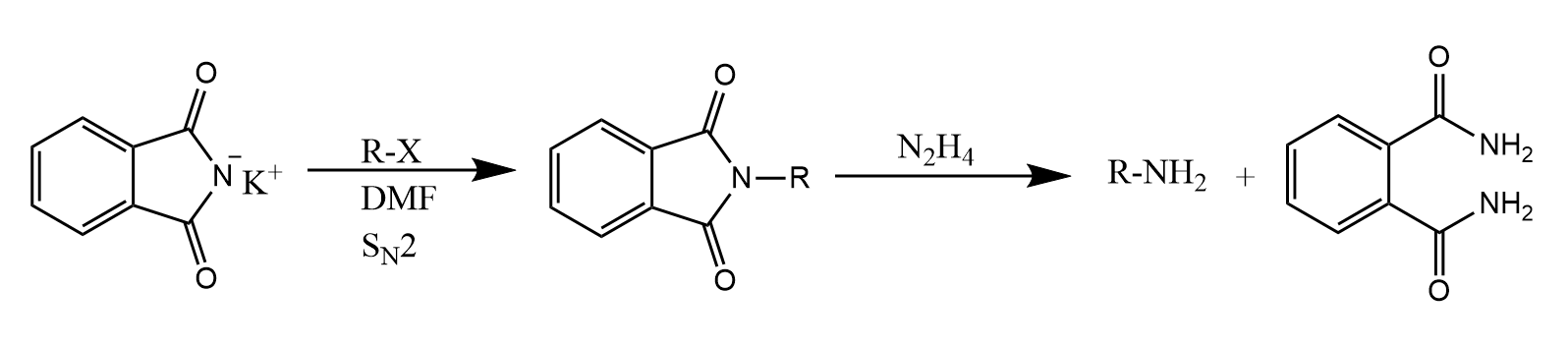
分子内亲核取代反应用表示,例如醇与二氯亚砜作用生成卤代烷的反应若在无溶剂条件下进行,反应则按照反应进行:
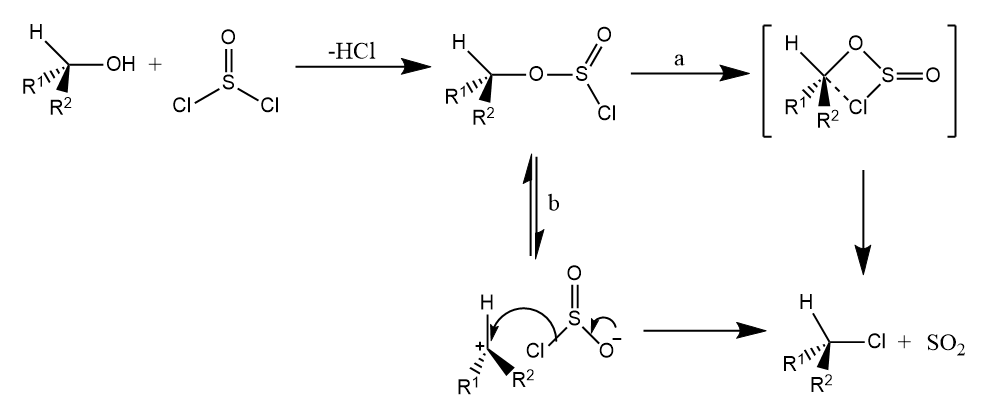
反应中可能经过协同的四元环过渡态,或经历离子对机理,这两种情况目前仍尚处争议。
邻近基团参与(简称邻基参与,NGP)是分子内富电子性中心对缺电子性中心进行捕获的一种现象,它和分子间反应相竞争,能改变反应速率、发生骨架重构、控制反应的立体化学。
发生邻基参与的前提是富电子性中心和缺电子性中心的轨道能级要匹配、轨道之间有足够的重叠。这些满足条件的基团先发生分子内亲核取代生成环状中间体,然后再接受亲核试剂的进攻,从而加快取代反应的速率。
羧基的氧原子容易发生邻基参与,比如下面这个很经典的例子:
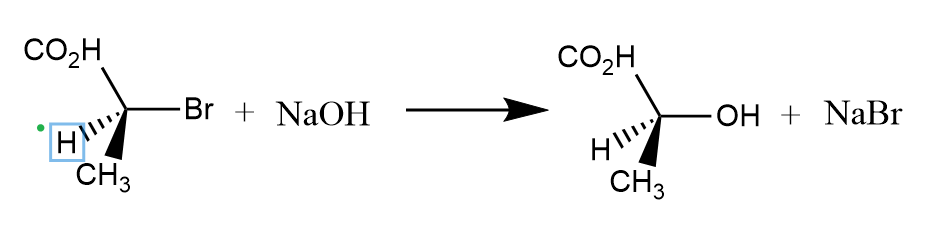
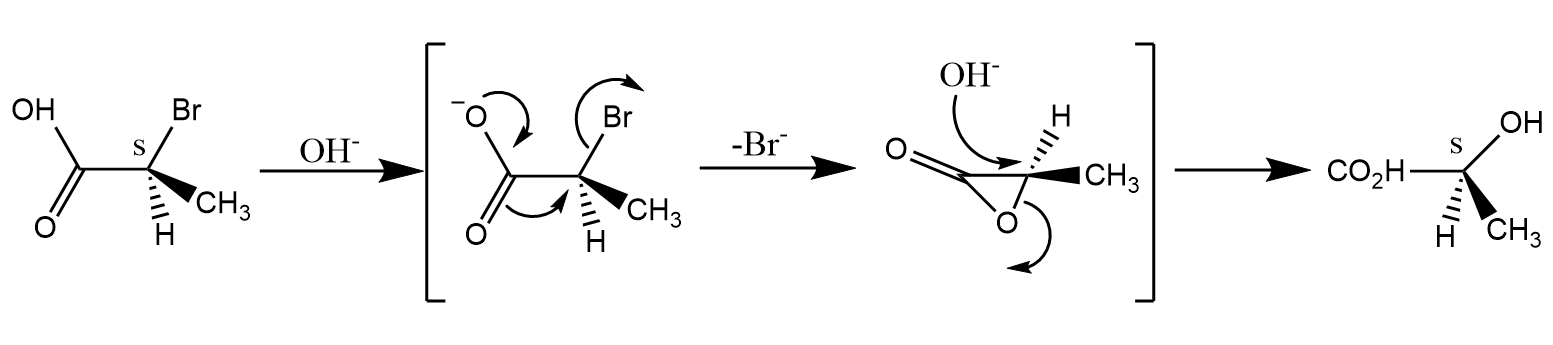
在该反应中,羧基负离子先发生邻基参与形成三元内酯中间体,然后亲核试剂再开环进攻,由于经历了两次反应,所以中心碳原子的构型得以保持。
类似的,酯基的氧原子亦容易发生邻基参与效应,如:
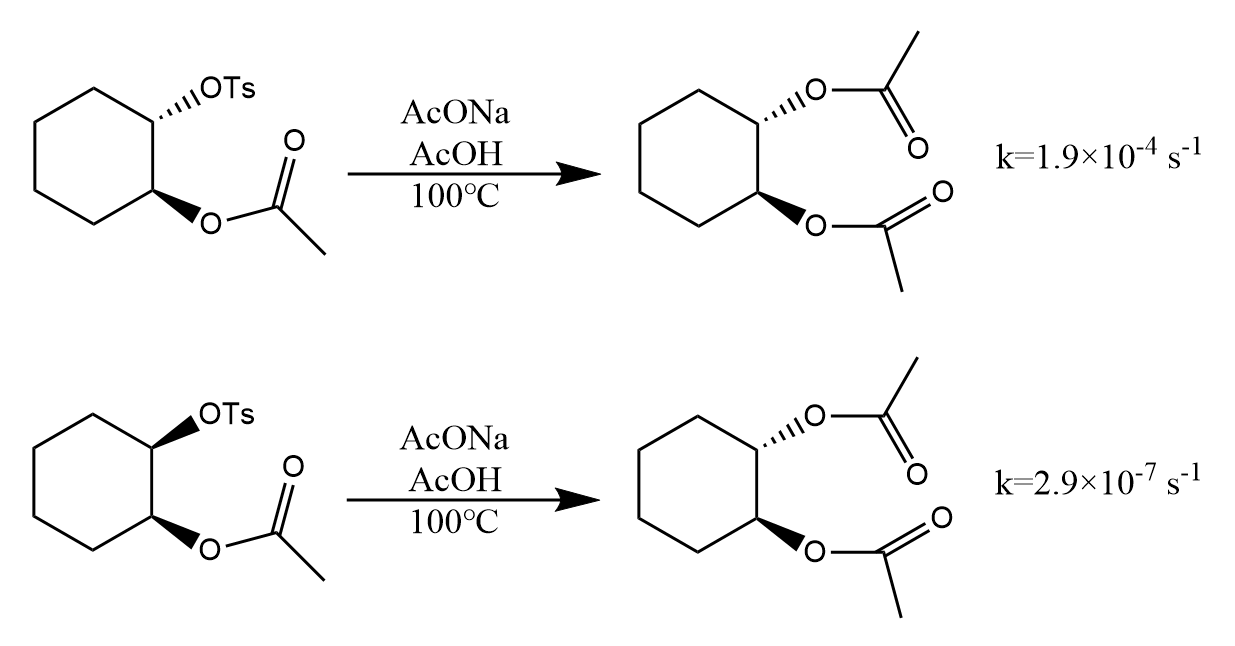
这两个反应都给出反式产物,第一个反应为反式底物,立体构型上易于发生邻基参与,所以最终构型保持;第二个反应为顺式底物,难以发生邻基参与,通过机理以相同的五元环氧鎓中间体完成反应。
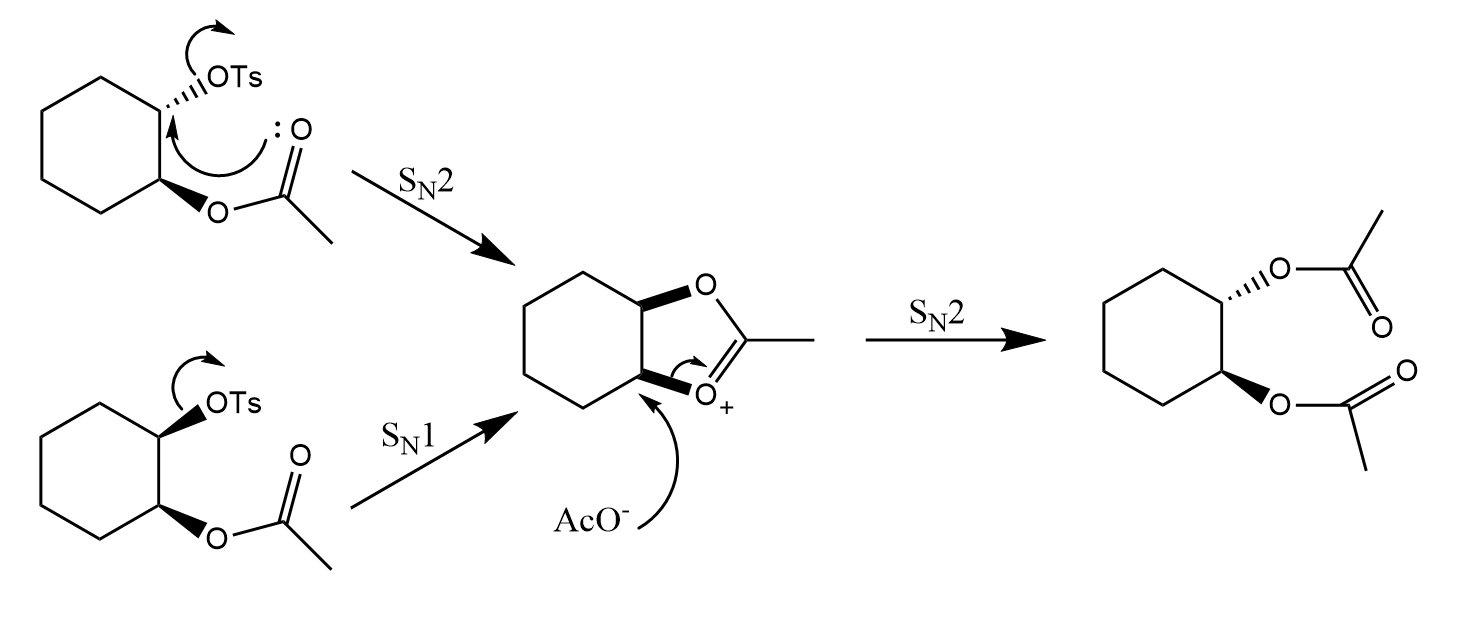
与氧相比,硫的亲核性更强,也就更易于发生邻基参与。
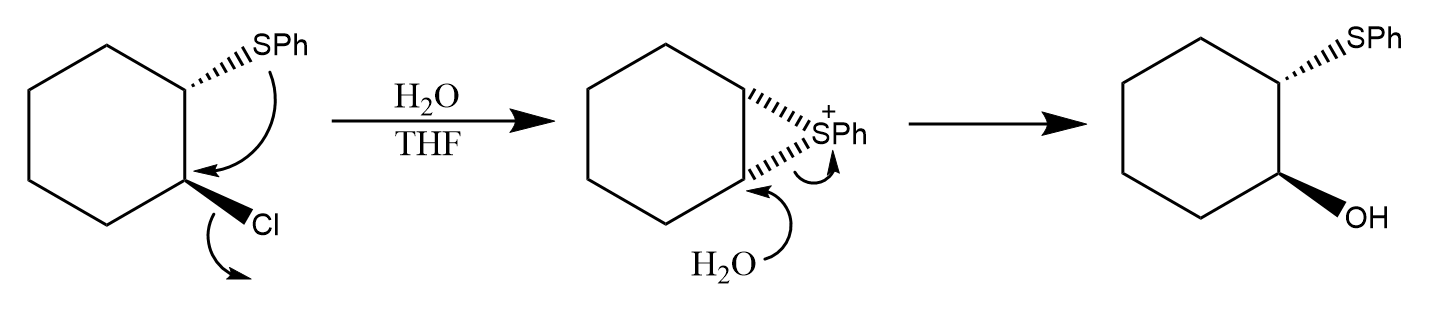
氮也可以发生邻基参与,如2-氯甲基-N-乙基吡咯烷能够通过氮的邻基参与发生扩环:
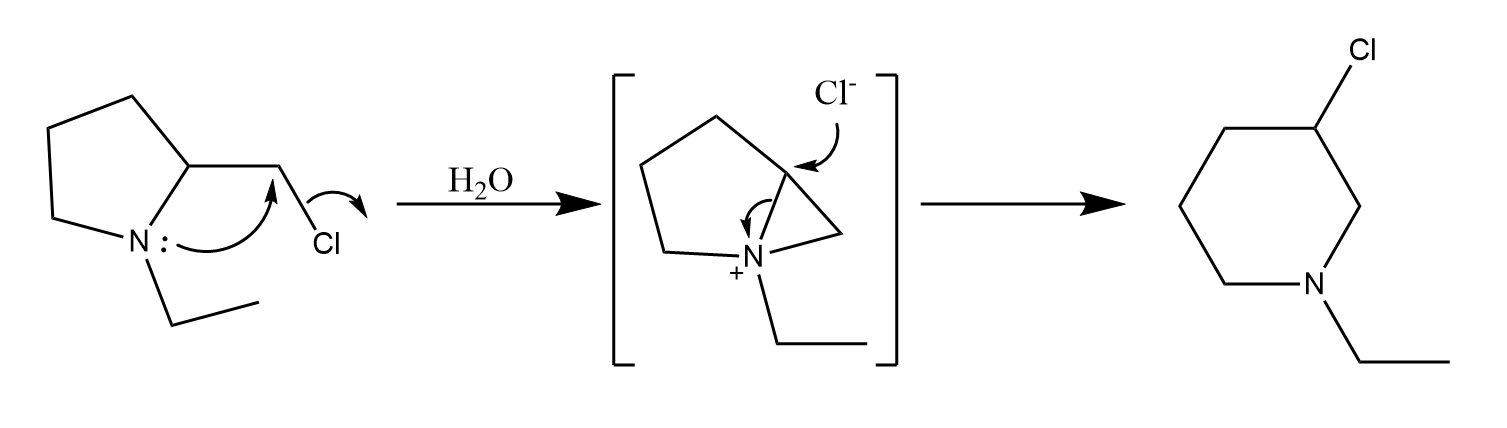
具有6π电子体系的苯环是常见的邻基参与基团,苯环的邻基参与经历了一个螺环结构的苯鎓离子中间体。
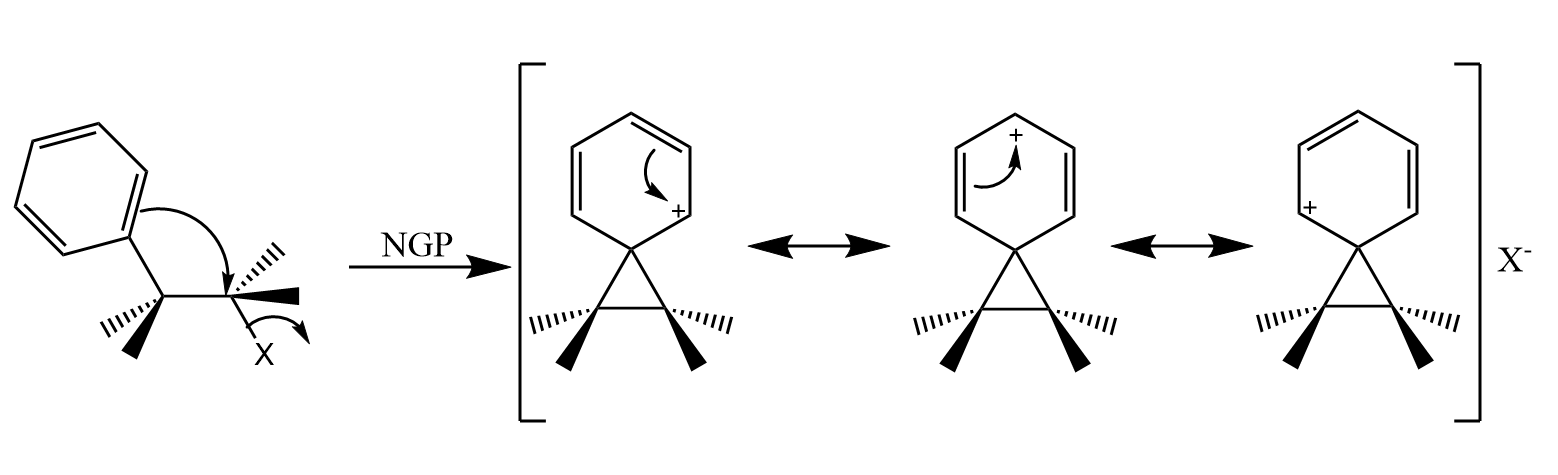
苯环的这种邻基参与效应已经得到了立体化学研究结果的支持,试验结果与猜测机理相符。既然邻基参与的基团是电子给体,那么基团的富电子性越强,其邻基参与就越显著。给电子基能够稳定苯鎓离子中间体,降低过渡态势能,有效地促进邻基参与的发生:
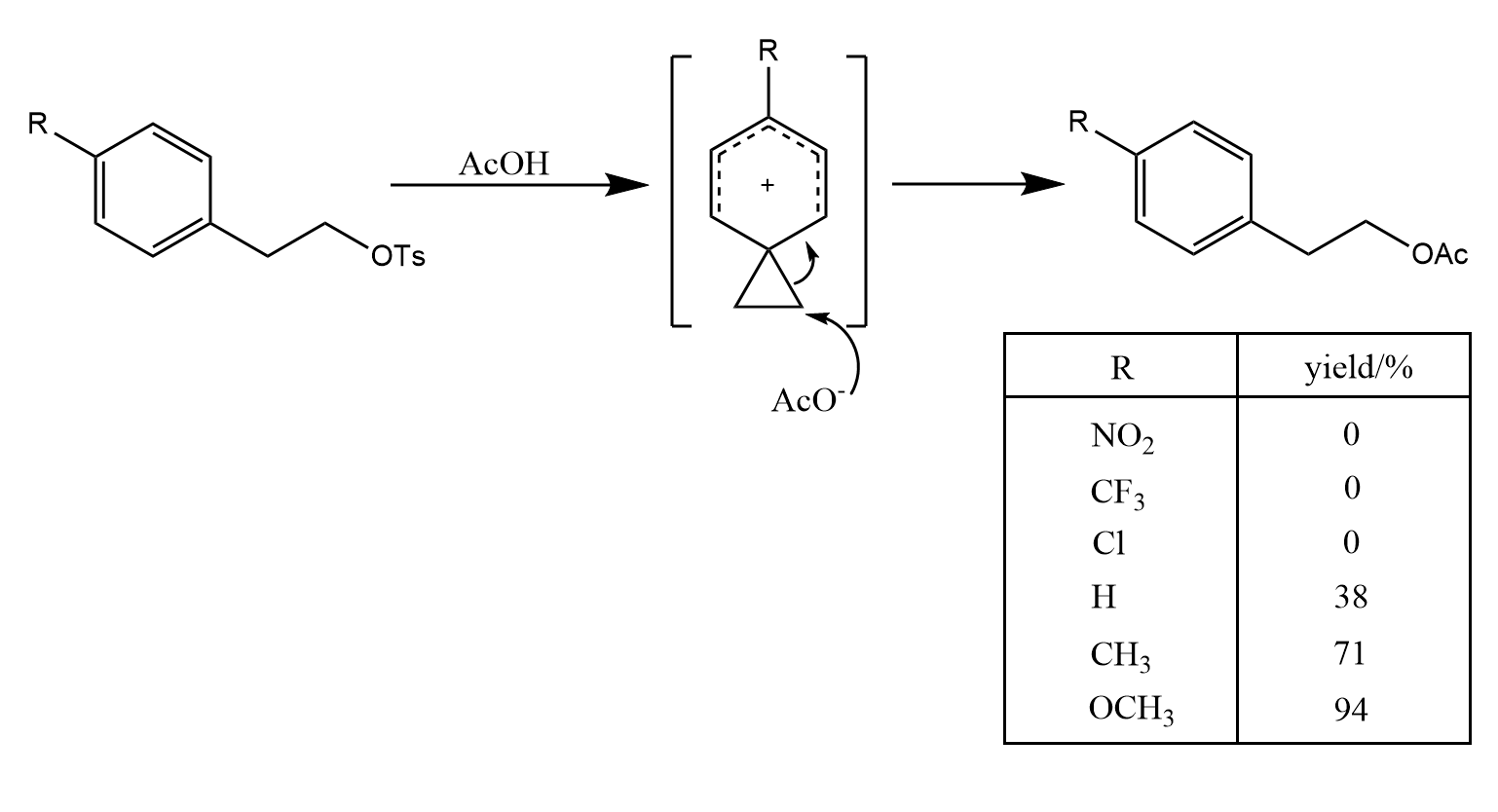
烯烃能够提供π电子而产生邻基参与效应,反应经历一个非经典的碳正离子中间体:
在上一个例子中,具有双键的底物(Ⅰ)比底物(Ⅱ)的反应快倍。
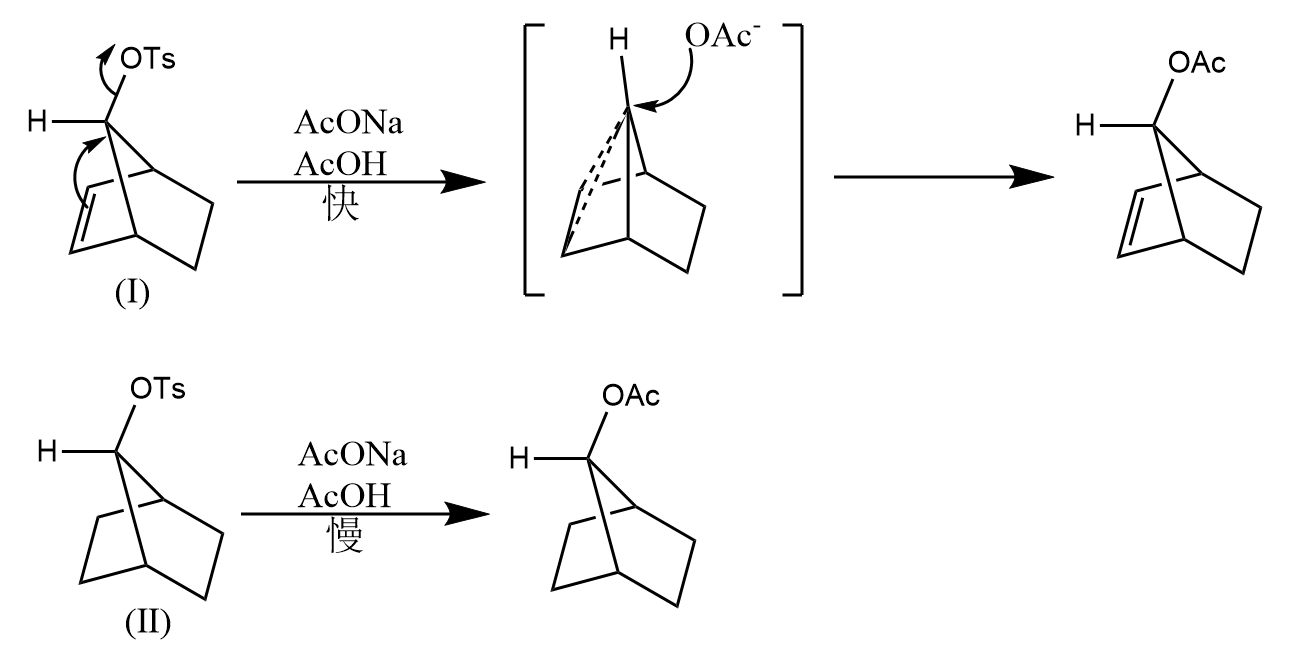
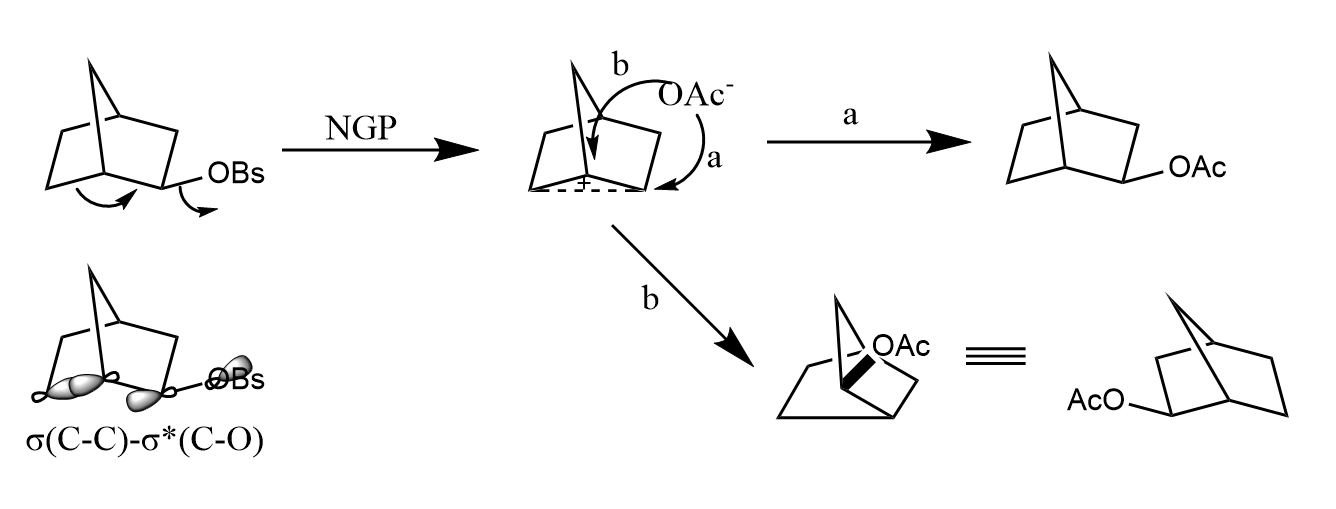
碳碳单键也能提供电子来促进亲核取代反应的发生,如下面的例子:
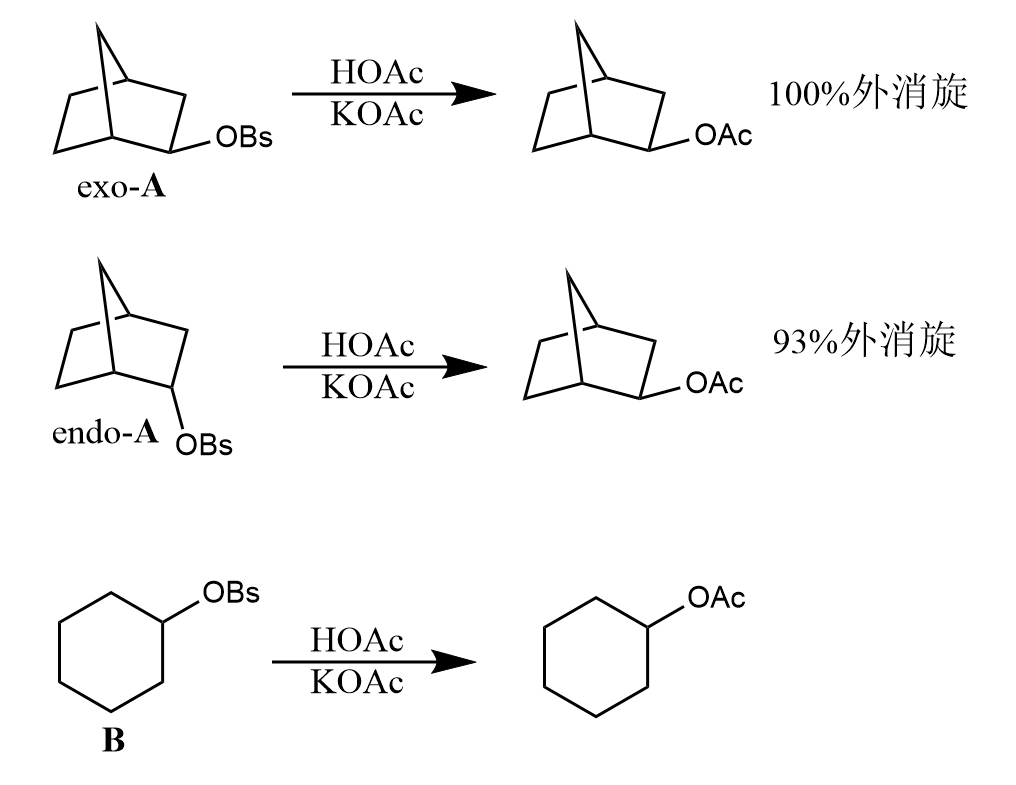
三种化合物的相对反应速率为。表明最易发生亲核取代,和的反应速率不受骨架影响。速率很快的原因在于分子内存在和之间的超共轭作用,能促进键解离,形成具有对称面的非经典碳正离子中间体,因此最终会得到100%外消旋产物。
6.2 芳环上的亲核取代反应
芳烃上的卤素不能接受亲核试剂的直接进攻,因为卤素和芳环存在一定共轭,键拥有部分双键性质,具有一定的稳定性。所以芳环上的亲核取代反应通常经过其他机理。
当芳香烃上有吸电子取代基时(EWG),亲核试剂对键发生亲核取代反应经过加成-消除机理。
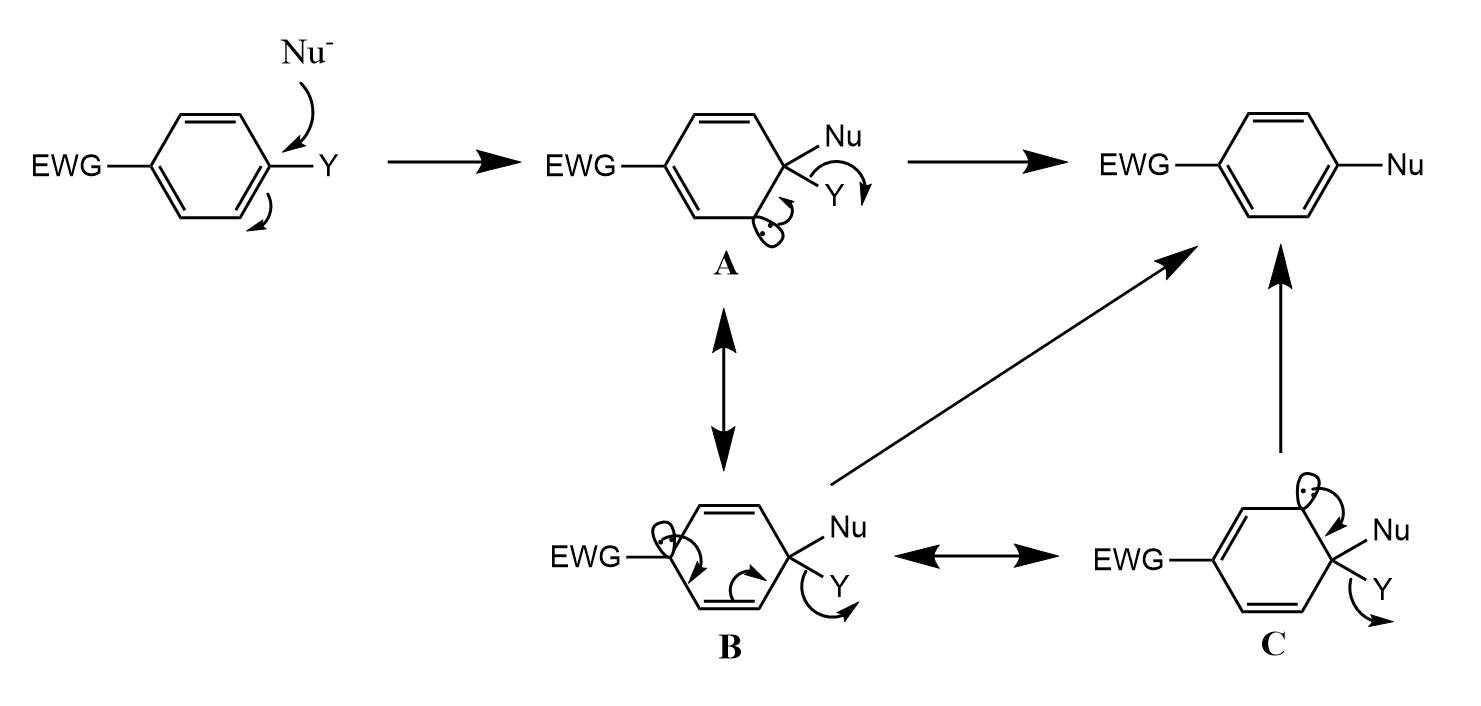
该机理的直接证据是分离得到中间体Meisenheimer盐:
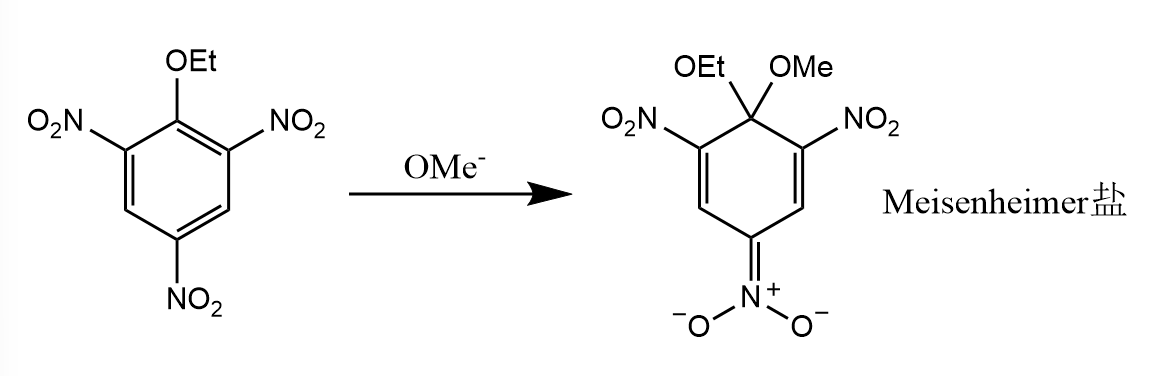
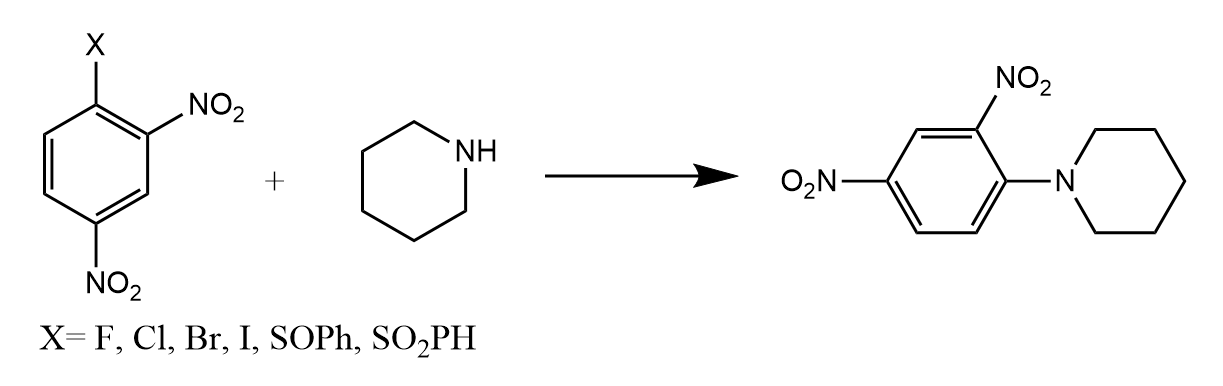
吸电子基的强度会显著影响反应的速率,很容易理解 ,芳环越缺电子,那么反应就越易发生。一个具体的例子:2,4-二硝基氟苯与哌啶的亲核取代反应速率是2,4-二硝基碘苯的3300倍,虽然碘离子是更好的离去基团,但是氟的吸电子能力更强,所以反应更快。
除了常见的离去基团,氢也可以离去。例如吡啶与氨基钠反应,通过加成-消除机理生成2-氨基吡啶,该反应称为Chichibabin反应:
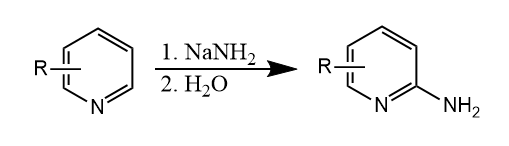
反应中经历氢负离子的消除,氢负离子在体系中立即反应生成氢气。
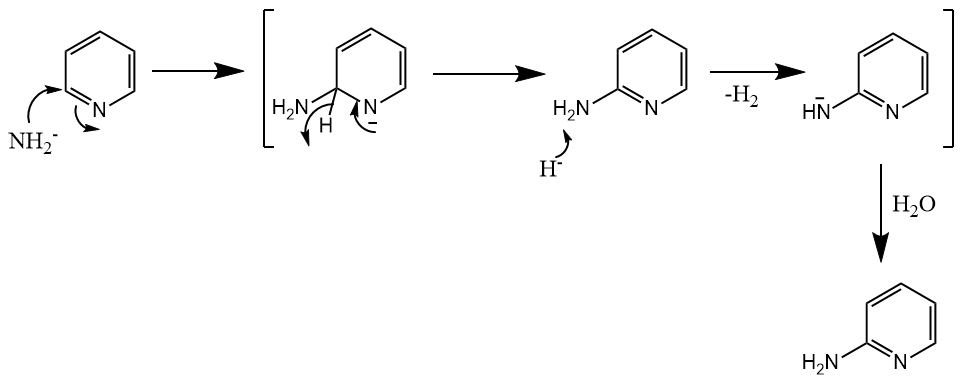
( 这个反应的名字莫名好笑,所以我印象比较深刻,Chichibabin是一位俄罗斯化学家的名字。另外该反应与下文提及的VNS有异曲同工之妙。 )
除了加成-消除机理外,芳香亲核取代还有消除-加成机理,这种机理会经历苯炔中间体,一般于反应过程中原位产生。
三氟甲磺酸(2-三甲硅基苯基)酯是常用的苯炔前体,它可在氟负离子的促进下生成苯炔:
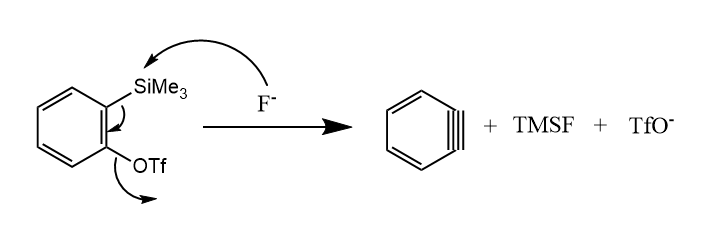
除此之外,邻氨基苯甲酸衍生的重氮盐的热消除、邻氟苯基格氏试剂的消除也是产生苯炔的重要方法。
苯炔非常活泼,许多亲核试剂都能与它发生亲核加成反应,产物相当于是亲核试剂对于苯炔的插入,例如苯炔与丙二酸二乙酯的反应:
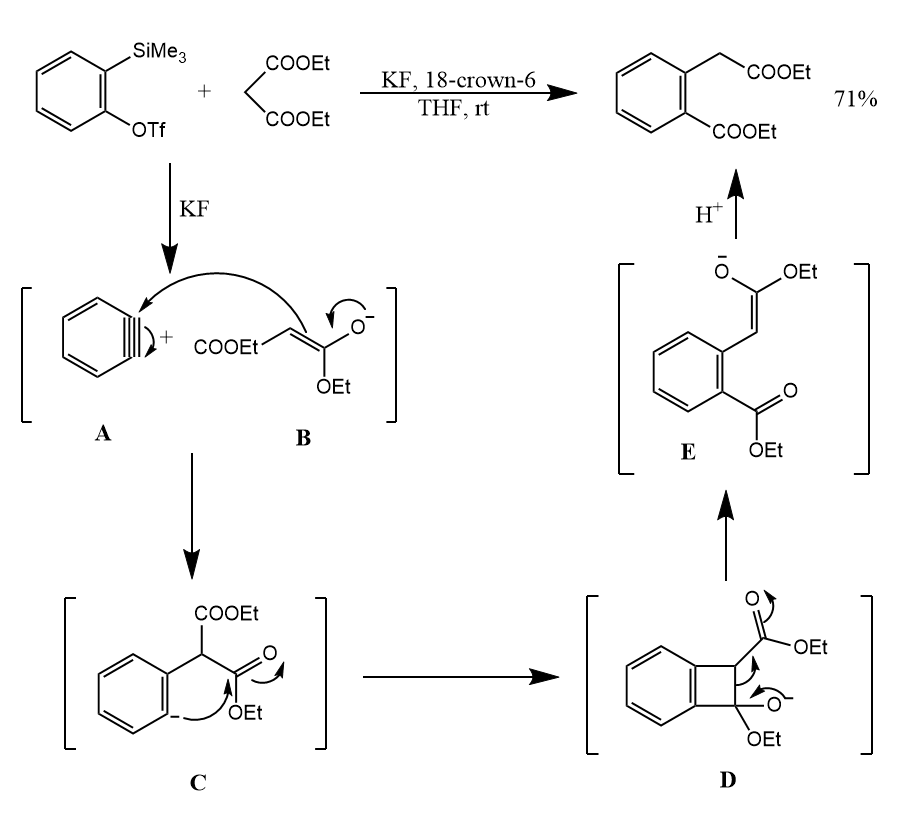
上述反应的净结果相当于苯炔对键的插入。下面是更多的例子:
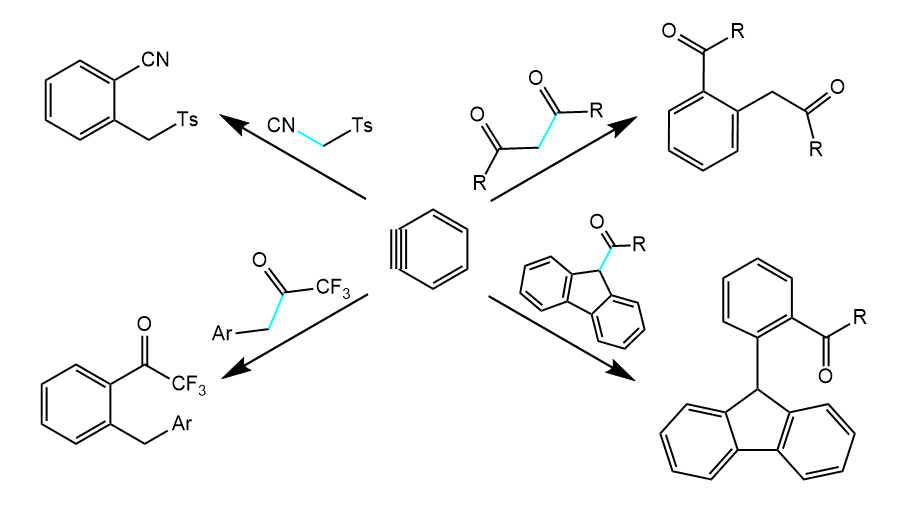
这种插入反应可以用于构建各种环,例如天然产物curvularin的合成:
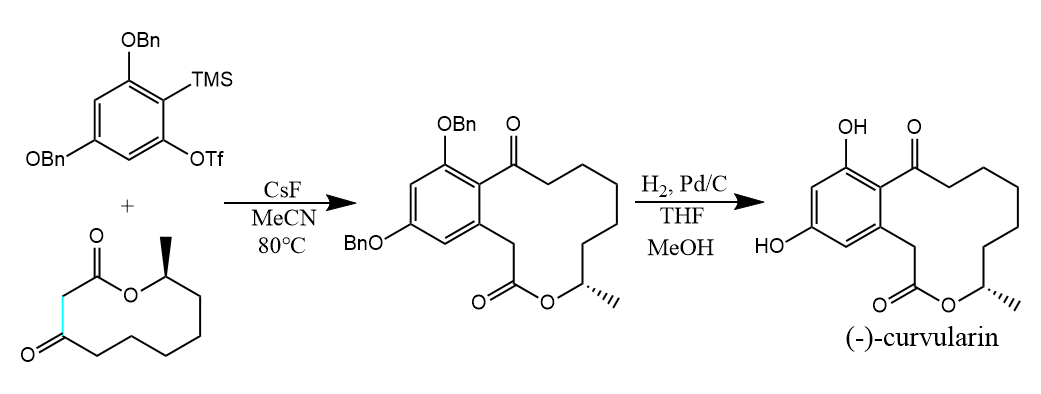
除键插入外,苯炔还可以发生键插入或键插入,例如苯炔对DMF的双键插入:
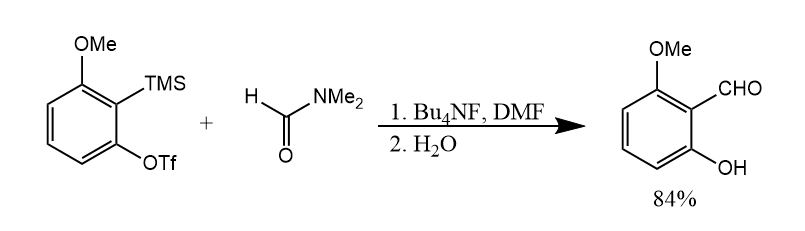
在该反应中,DMF中羰基氧亲核进攻苯炔形成芳基负离子,接着通过四元环的成环开环成为亚胺盐中间体,最后水解得到最终产物。若把捕获剂换为苯甲酰腈,酰基作亲电基团,氰基负离子作亲核基团,得到如下产物:
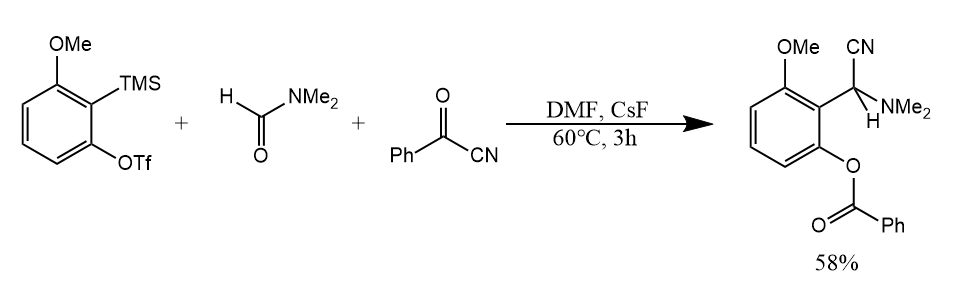
除了上述提到的常规苯炔前体外,一般的卤代物也可以发生消除-加成反应:
对于不同的卤代物,反应性为溴苯>碘苯>氯苯>氟苯,这个顺序是离去性和吸电子诱导效应的综合结果。
在苯炔的形成和反应过程中,取代基的诱导效应都产生重要影响,由于取代基的诱导效应,炔键的形成位置和亲核试剂的加成位置都会不同。例如间甲氧基氯苯的消除-加成反应:
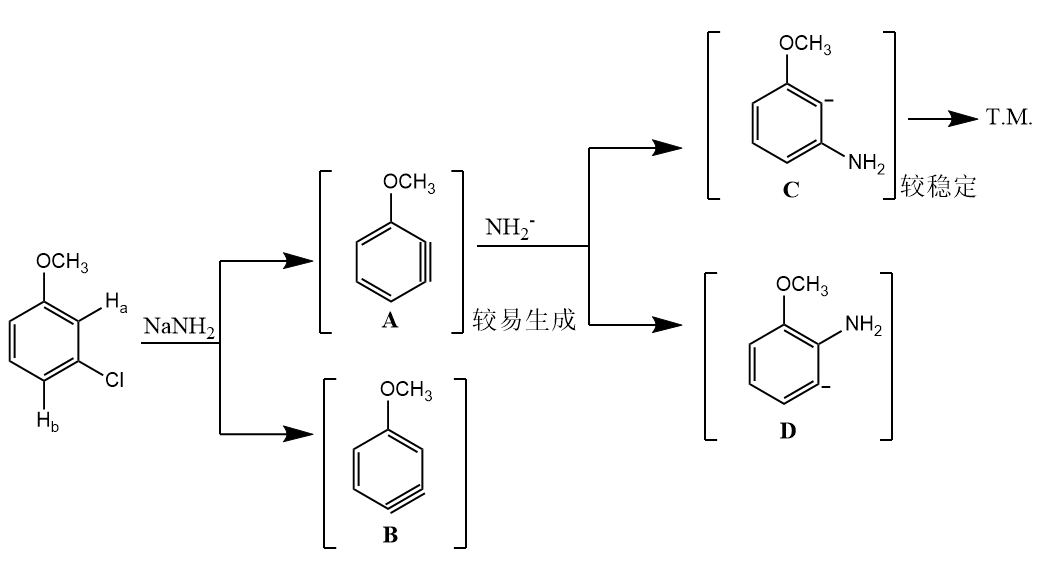
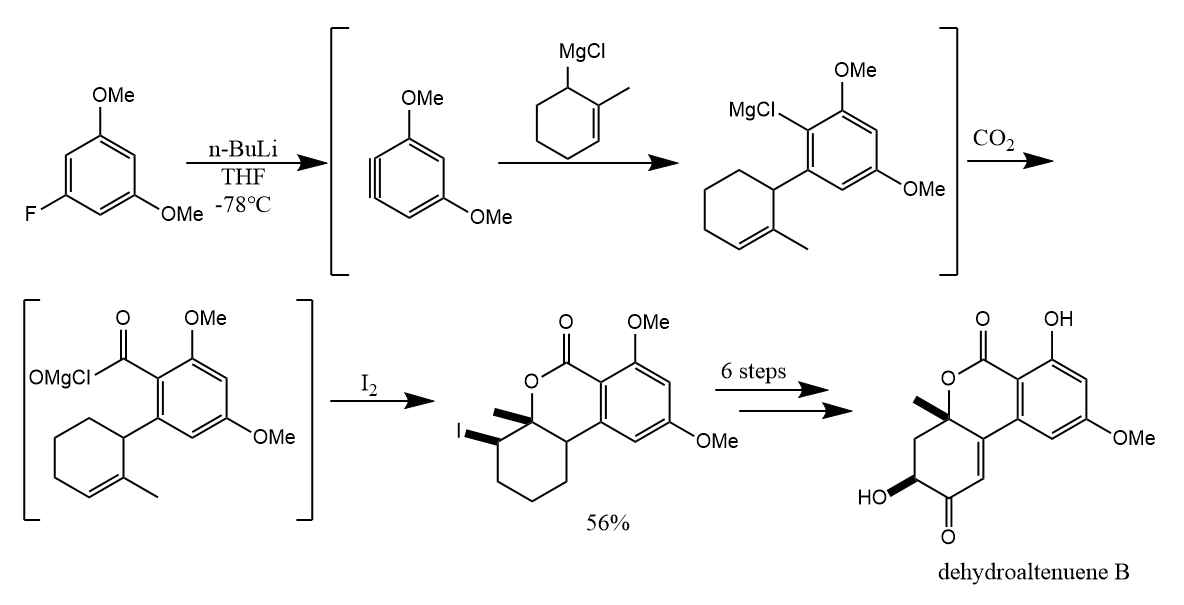
在消除-加成机理中,加成所产生的芳基负离子可进一步被亲电试剂捕获,完成一系列串联反应。例如天然产物dehydroaltenuene B的全合成:
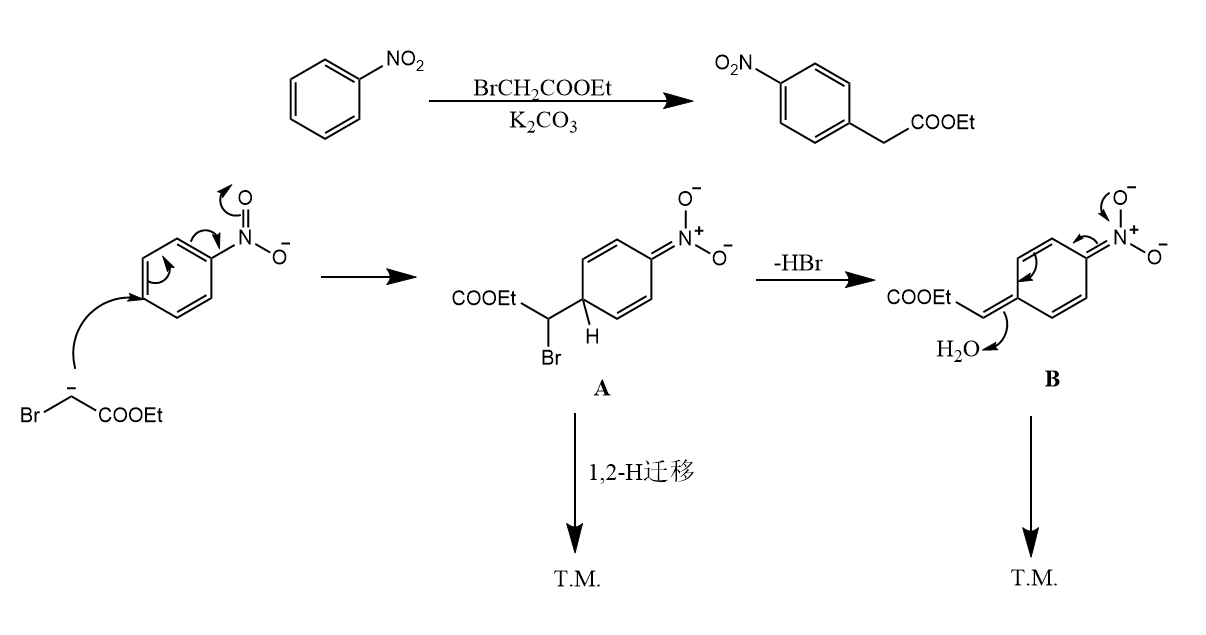
当缺电子性芳香烃和带有离去基团的亲核试剂反应时,生成芳香烃上氢被亲核取代的产物,该反应则称为芳香烃间接亲核取代(vicarious nucleophilic substitution, VNS)。该反应的关键是亲核试剂上自带的离去基团的离去,这一步可以通过协同的1,2-H迁移途径或在碱性条件下的消除途径完成:
芳香亲核取代还有一种芳基碳正离子机理,这种机理较为少见,最常见的是芳基重氮盐的反应,反应属于单分子亲核取代反应。例如Balz-Schiemann反应:
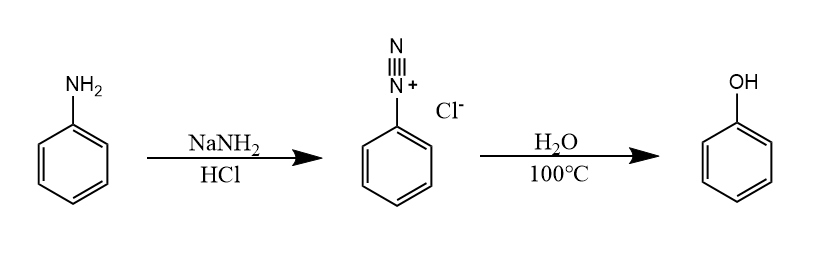
这是一个向芳环上引入氟原子的有效反应。
6.3 羧酸及其衍生物的亲核取代反应
亲核试剂进攻羧酸、酰氯、酸酐、酯或酰胺的羰基碳,经历加成-消除机理后得到亲核取代产物。这也是基础有机中经典的反应,反应活性为酰氯>酸酐>酯>酰胺。
羧酸衍生物之间可以互相转化,例如各衍射物水解则得羧酸,用醇解则得酯,氨解则得酰胺。这些反应的机理本质上都相同,都是先加成,后消除的途径,根据条件不同(酸/碱催化、单分子/双分子机理、酰氧键/烷氧键断裂)可以分为八种:。
当然以上只是排列组合的结果,实际上尚未通过实验证实,另有几种机理也极为少见。最常见的两种机理为四面体机理:和。
本节中许多反应都较为常见,在此不多赘述,仅讨论酰胺活化内容。
酰胺因其结构中的共振,使其性质很稳定,不易发生亲核取代反应,调整胺的结构可以有效抑制酰胺的共振:
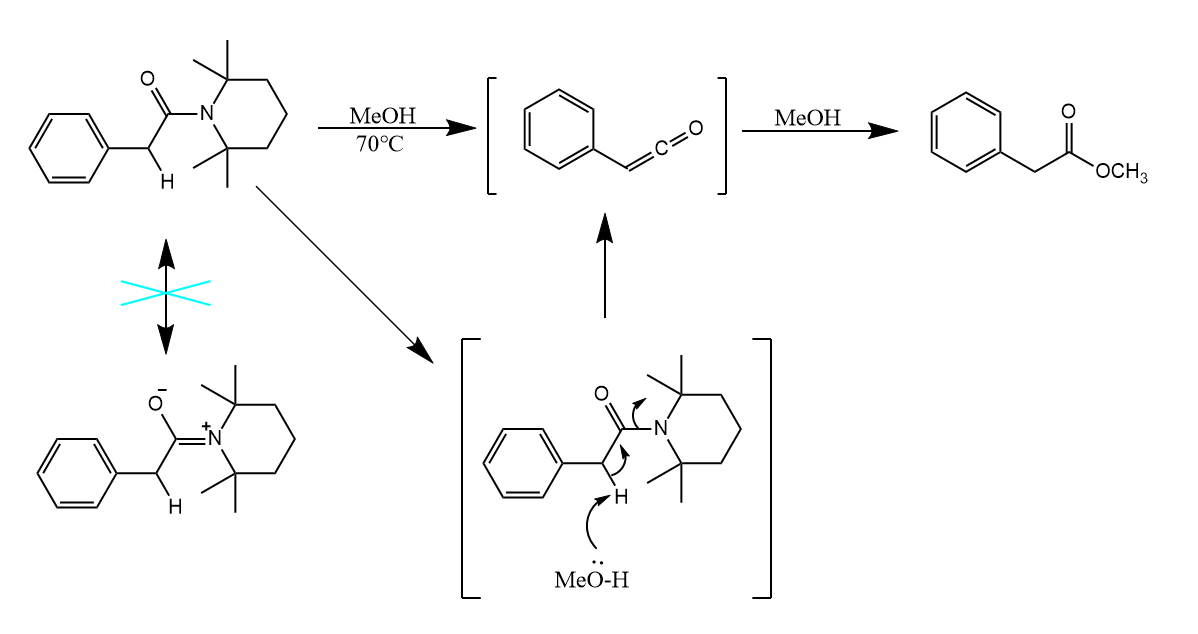
在上例中,底物四个甲基的位阻阻碍了酰胺的共振,因而使得转化活性提升。
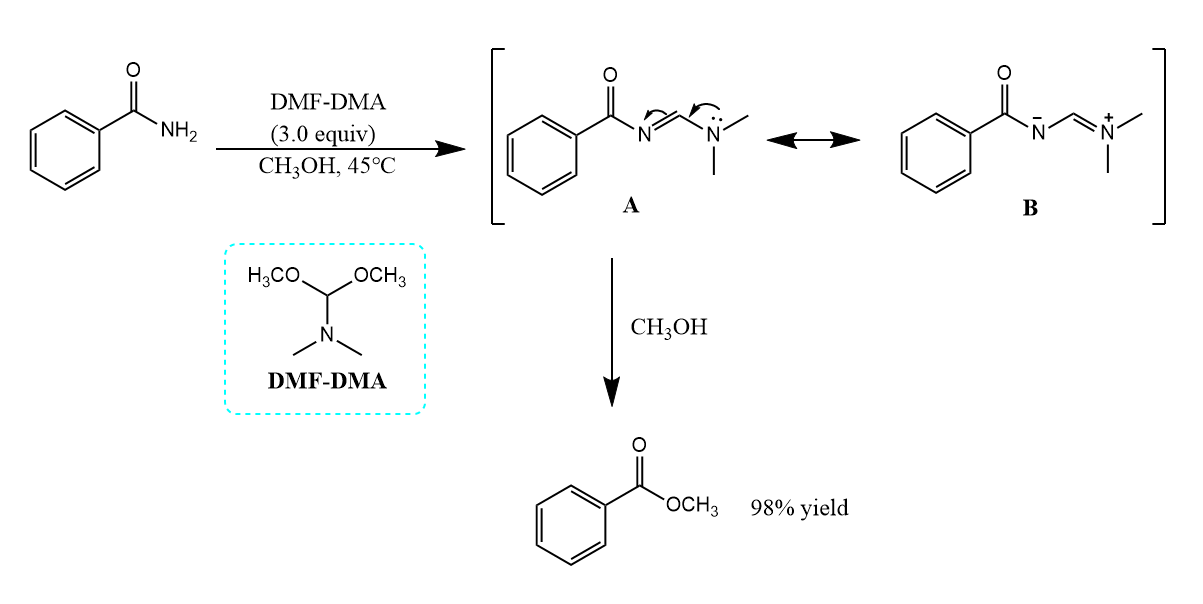
也可以让酰胺的N端形成稳定结构来削弱酰胺的共振,例如:
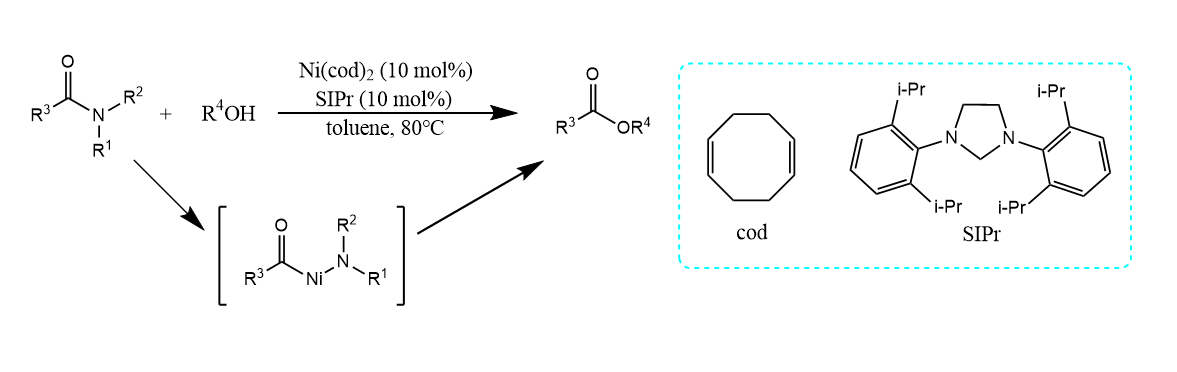
最直接的方法还是利用过渡金属直接活化并切断酰胺的键:
总结
本章为亲核取代反应,邻基参与的部分是之前了解不多的内容。
悲伤画图…