《ART》章末总结4
《ART》第四章,开始周环反应部分,前三章都是只写了Summary部分,因为基本反应在基础有机里都还学得较多,理论和反应看着都亲切一些。不过从周环反应开始就更陌生一些( 因为我周环本身就学的挺烂 ),所以开始整理一下正文的内容。就不作翻译了,权当学英语吧XD。
4.1 Introduction
A pericyclic reaction is a reaction in which bonds are formed or broken at the termini of one or more conjugated $\pi$ systems. The electrons move around in a circle, all bonds are made and broken simultaneously, and no intermediates intervene.
For each class of pericyclic reactionss two or more of the following characteristics will be discussed: the typical reactions, regioselectivity, stereoselectivity and stereospecificity.
4.1.1 Classes of Pericyclic Reactions
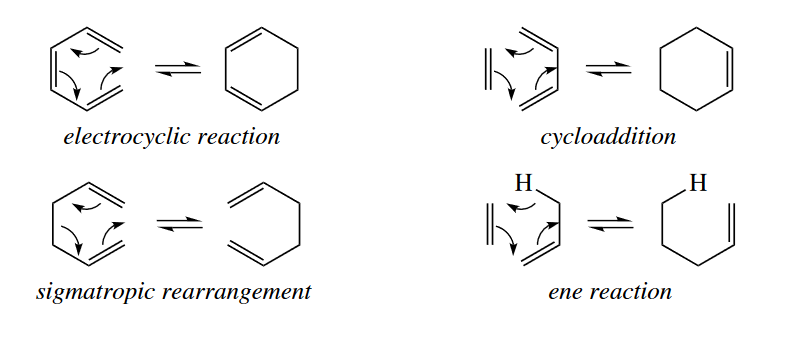
There are four major classes of pericyclic reactions: electro-cyclic reactions (ring openings or ring closings), cycloadditions, sigmatropic rearrangements, and ene reactions.
In an electrocyclic ring closing, a $\sigma$ bond forms between the termini of a conjugated $\pi$ system. An electrocyclic ring opening is the reverse reaction.
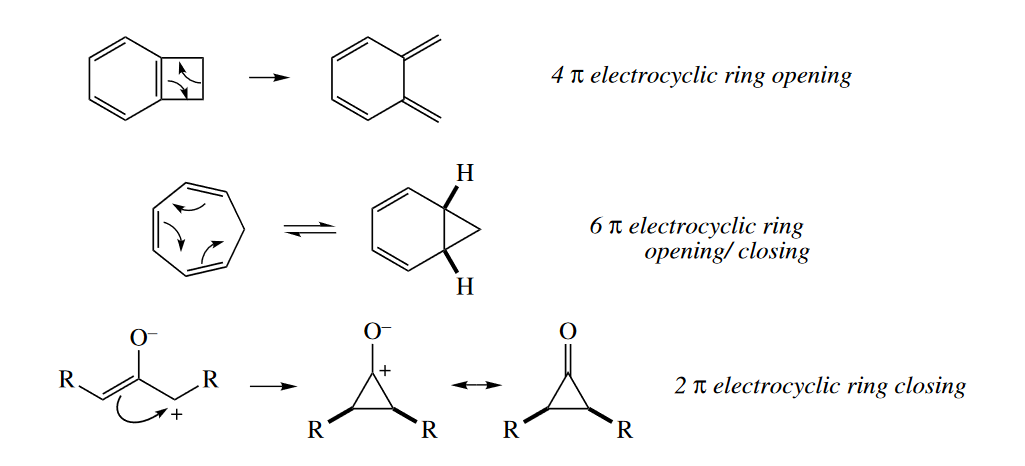
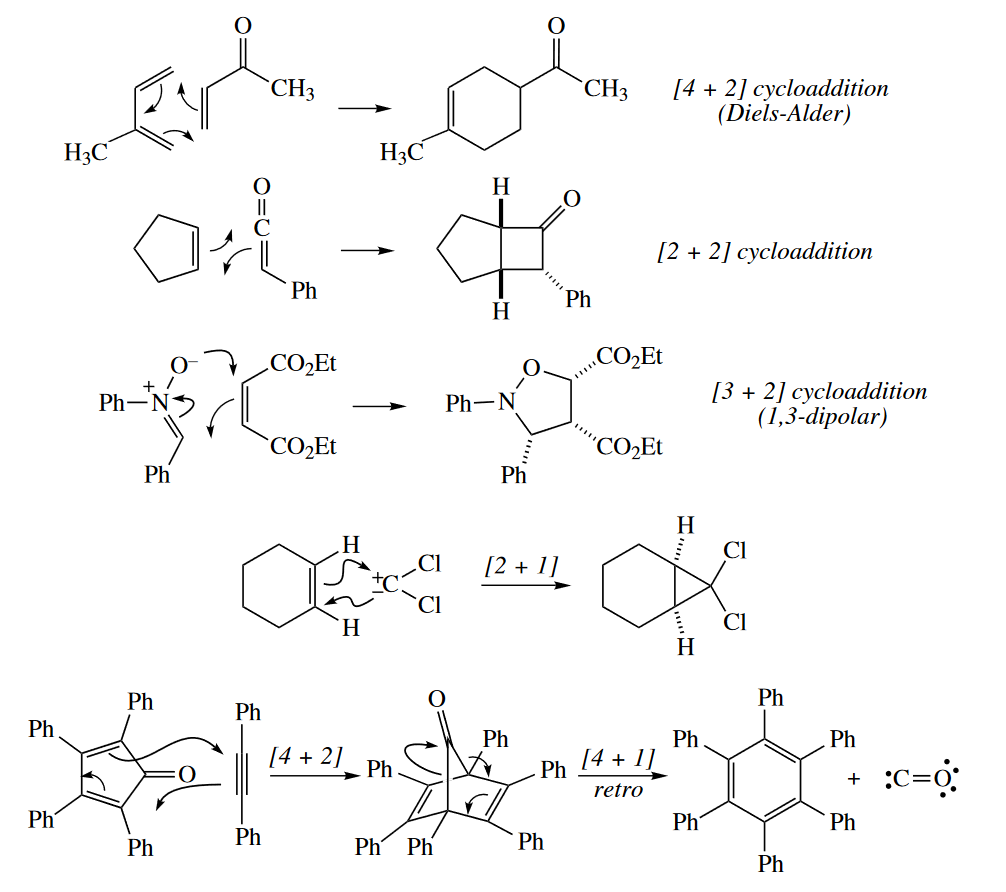
When $\sigma$ bonds are formed between the ends of two $\pi$ systems to give a cyclic product, the reaction is called a cycloaddition. The reverse reaction is called a retro-cycloaddition. Cheletropic reactions are a special class of cycloadditions in which one of the components is a single atom. The one-atom component must have one full and one empty orbitals; it may be a carbene, $SO_2$, or $CO$.
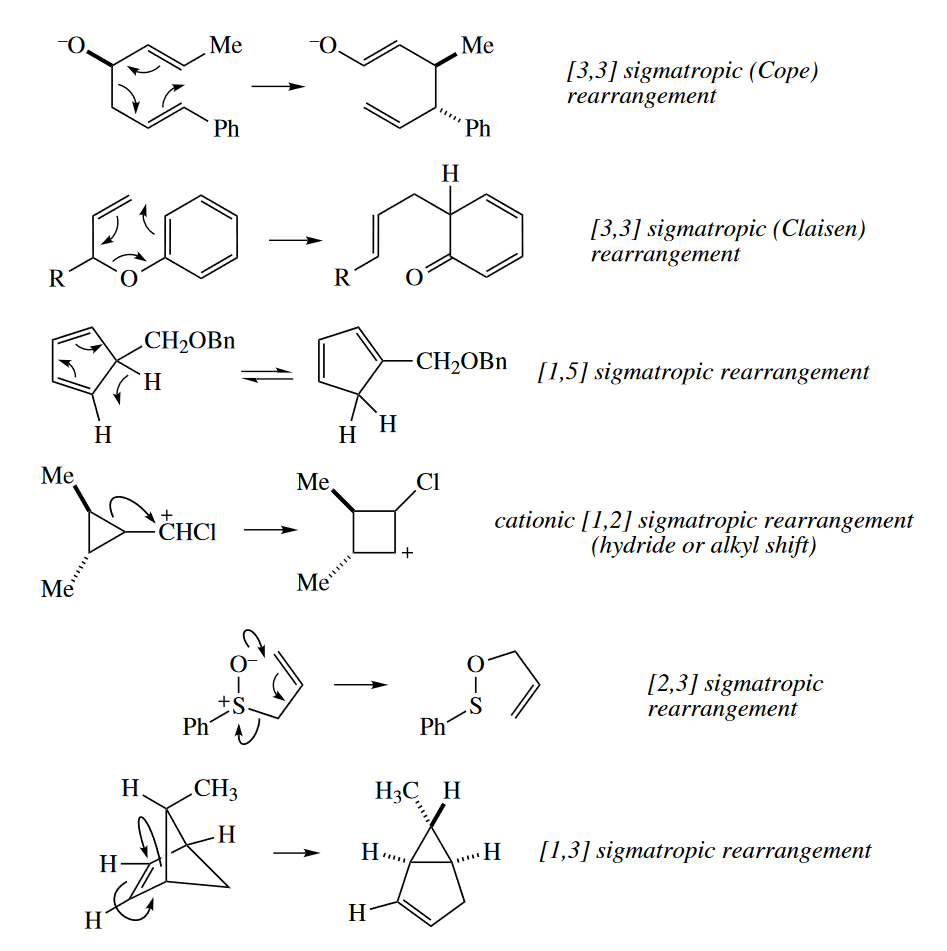
Sigmatropic reaarrangements involve the cleavage of a $\sigma$ bond connecting the end of one fragment with the end of another, and concerted formation of another $\sigma$ bond at the other ends of the fragments. The $\sigma$ bond seem to migrate, hence the name of the reaction.
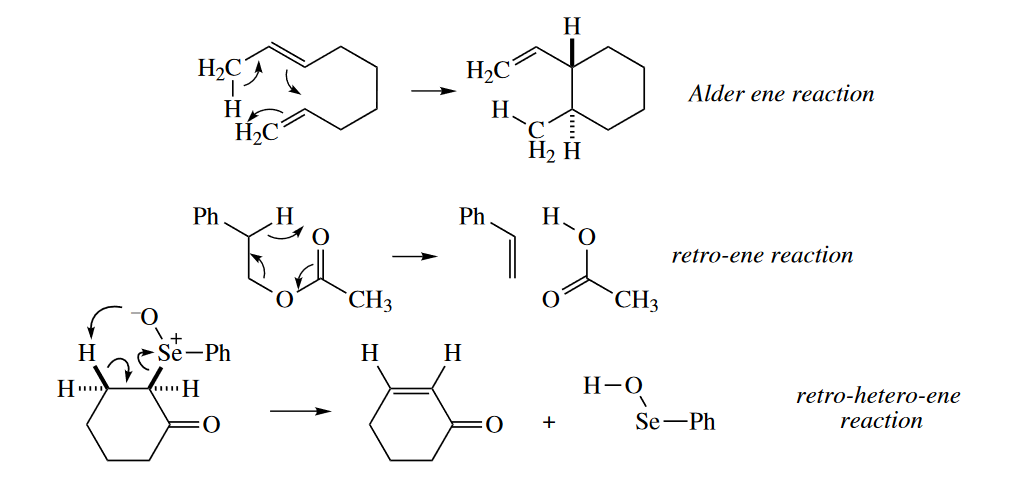
The ene reaction is always a six-electron reaction. It shares some characteristics with the [4+2] cycloaddition and the [1,5] sigmatropic rearrangement. Like the [4+2] cycloaddition, a four-atom component reacts with a two-atom component, but in the ene reaction, the four-atom component consists of a $\pi$ bond and an allylic $\sigma$ bond (usually to a H atom), not two $\pi$ bonds. Like a [1,5] sigmatropic rearrangement, a $\sigma$ bond migrates, but the ene reaction also produces one $\sigma$ bond at the expense of one $\pi$ bond.
Archetypical six-electron examples of each class of pericyclic reaction are shown in the following figure.
Stereospecificity, the property that the stereochemistry of the starting materials determines the stereochemistry of the product, is one of the hallmarks of pericyclic reactions. It is possible to draw two-step nonconcerted, polar or free-radical mechanisms for many pericyclic reactions, but these two-step mechanisms fail to account for the stereospecificity of the reactions.
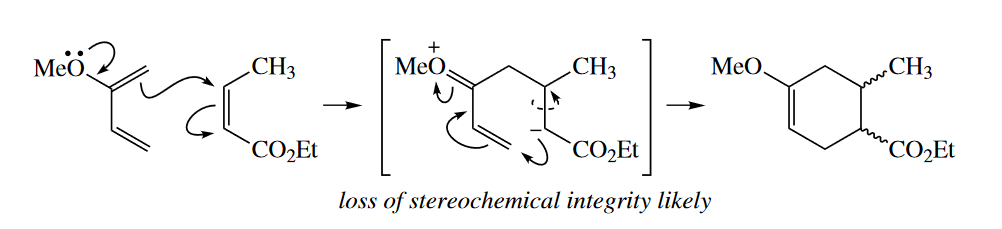
( 这是很重要的一个判断方法,周环反应一般具有特定的立体选择性,而普通的极性机理或自由基机理很难完全符合反应表现出的立体化学。 )
Like all reactions, pericyclic reactions are reversible in principle (even though they may be irreversible in practice). The forward and reverse reactions always go through the same transition state
4.1.2 Polyene MOs
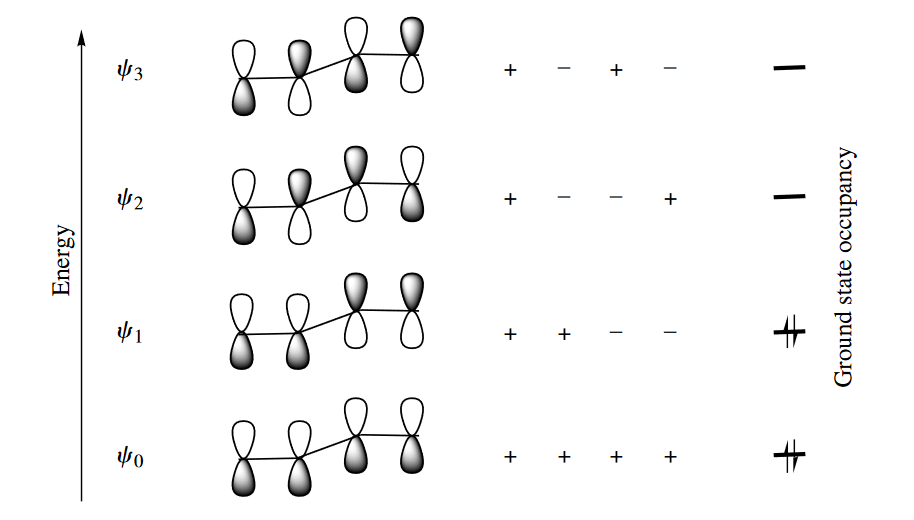
Consider 1,3-butadiene. Four p orbitals interact, each with its neighbors, to produce four MOs. These four orbital are called $\psi_o,\psi_1,\psi_2$ and $\psi_3$. In the lowest energy MO, $\psi_0$, every p orbital overlaps constructively with its neighbor to gibe an MO in which there are no nodes, or changes in sign betwwen orbitals. In the highest energy MO, $\psi_3$, every p orbital overlaps destructively with its neighbor to give an MO in which there are three nodes. In the intermediate energy MOs, $\psi_1$ and $\psi_2$, there are one and two nodes, respectively, and the MOs are constructed accordingly. Under thermal conditions, the four electrons derived from the four AOs occupy $\psi_0$ and $\psi_1$; $\psi_1$ is the HOMO, and $\psi_2$ is the LUMO.
In 1,3,5-hexatriene, six AOs produce six MOs, the signs of whose termini alternate from like, to opposite, to like, etc. The number of nodes increases with each MO. In the ground state of 1,3,5-hexatriene, $\psi_2$ is the HOMO and $\psi_3$ is the LUMO. Note that in $\psi_3$ and $\psi_4$, the p orbitals of the second and fifth C atoms of the chain have a coefficient of zero, indicating that the p orbitals from those C atoms do not contribute at all to $\psi_3$ and $\psi_4$. Two of the nodes in both $\psi_3$ and $\psi_4$ pass through the centers of these two C atoms.
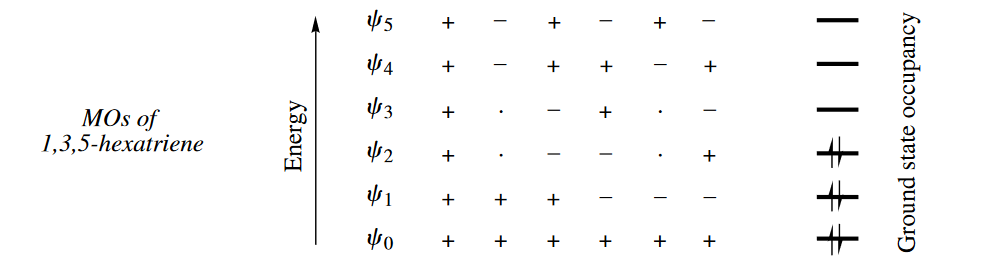
The symmetries of the MOs of conjugated $\pi$ systems with odd numbers of atoms also alternate. The allyl system has three MOs: $\psi_0$(symmetric), $\psi_1$(antisymmetric), and $\psi_2$(symmetric). In the ally cation, $\psi_0$ is the HOMO and $\psi_1$ is the LUMO, whereas in the allyl anion, $\psi_1$ is the HOMO and $\psi_2$ is the LUMO. The pentadienyl system has five MOs. In the pentadienyl cation, $\psi_1$(antisymmetric) is the HOMO and $\psi_2$(symmetric) is the LUMO, whereas the pentadienyl anion, $\psi_2$ is the HOMO and $\psi_3$(antisymmetic) is the LUMO.

( 条件不同会导致共轭多烯具有不同的电子排布,因而影响HOMO和LUMO,进而影响反应的立体化学。
在原子轨道线性组合为分子轨道时,每个原子轨道对新形成的分子轨道的贡献并非相等,而是有不同的组合系数,轨道系数的大小提示了反应性的高低。 )
4.2 Electrocyclic Reactions
4.2.1 Typical Reactions
Cyclobutenes are in electrocyclic equilibrium with 1,3-butadienes. 1,3-Butadienes are lower in energy, because cyclobutenes are very strained, and it is therefore possible to convert a cyclobutene to a butadiene thermally. The reverse reaction, the conversion of a 1,3-butadiene to a cyclobutene, does not usually proceed under thermal conditions, as it involves an uphill climb in energy. However, the more conjugated 1,3-butadienes absorb light at longer wavelengths than cyclobutenes, so it is possible to convert a 1,3-butadiene to a cyclobutene photochemically by choosing a wavelength at which the butadiene absorbs and the cyclobutene does not.
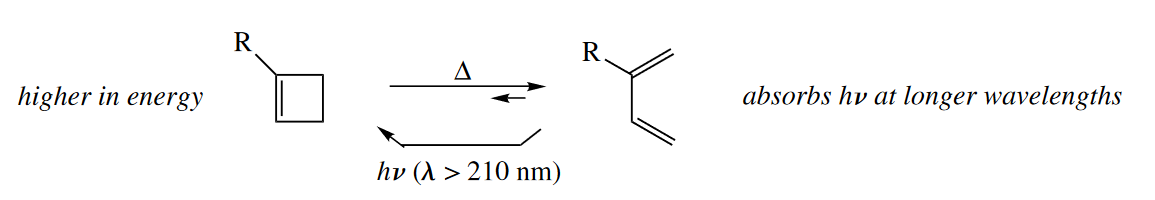
1,3-Cyclohexadienes are in electrocyclic equilibrium with 1,3,5-hexatrienes. Neither compound is strained, and the cyclohexadiene has one more $\sigma$ bond than the hexatriene, so the cyclohexadiene is lower in energy. The hexatriene is more conjugated than the cyclohexadiene, so it is more reactive photochemically. Under both thermal and photochemical conditions, then, the cyclohexadiene is favored over the hexatriene.
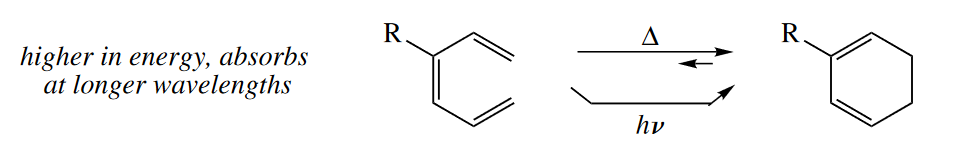
Allyl and pentadienyl cations participate in electrocyclic ring openings and closings, too. (The corresponding anions undergo similar reactions, but they are less important synthetically.) Less highly conjugated cations are produced upon ring closure of these species, but the decrease in cation stabilization is compensated by the gain of a C–C $\sigma$ bond. In the case of the allyl system, the increase in strain upon ring closing must also be considered.
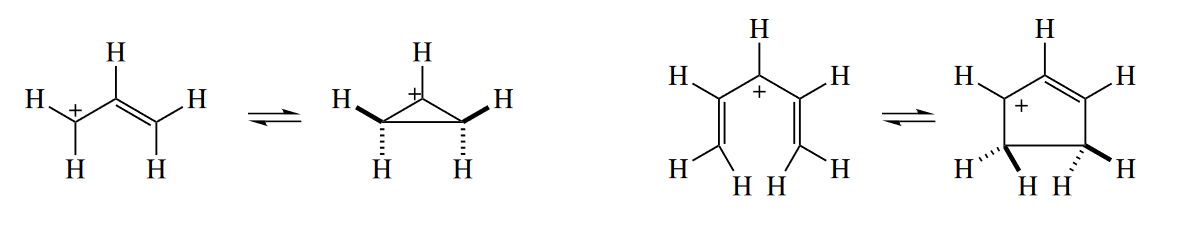
( 总结来说,电环化的关环产物通常更稳定,因为$\pi$键转化为$\sigma$键,但是如果关环产物的小环具有很大的环张力,那么也有可能让开环反应更易进行。在光激发条件下,多烯烃结构更易发生反应。 )
The electrocyclic equilibrium between cyclopropyl cations and allyl cations normally favors the allyl cation because the cyclopropyl cation is both more strained and less conjugated than the starting allylic cation. For example, the cyclopropyl halide shown undergoes concerted loss of $Br^-$ and electrocyclic ring opening to the allyl cation upon distillation. (In principle, a ring opening that is not concerted with loss of the leaving group could be drawn, but the cyclopropyl cation is very high in energy.) Addition of $Br^-$ to the allylic cation then gives the product.
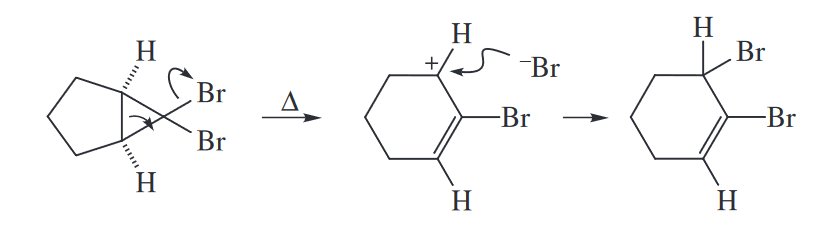
The carbene derived from halogen–metal exchange between RLi and a dibromocyclopropane can also undergo a two-electron electrocyclic ring opening to give an allene.
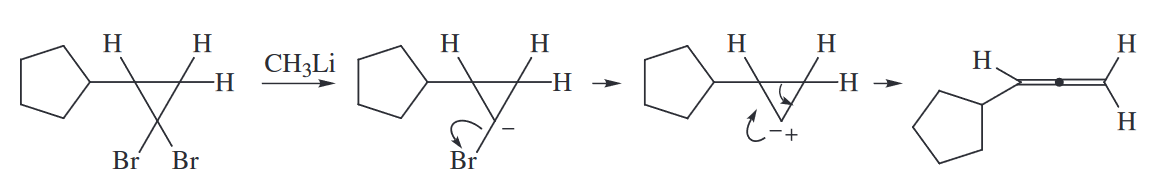
The key to identifying an electrocyclic ring closing is to look for the formaion of a new bond at the ends of a conjugated $\pi$ system. The key to identifying an electrocyclic ring opening is to look for the cleavage of a $\sigma$ bond joining the allylic positions of a conjugated $\pi$ system. Three-membered rings are closed from or opened to allylic systems by electrocyclic reactions.
4.2.2 Stereospecificity
In the electrocyclic reaction, the termini of the butadiene must rotate in order for the p orbitals at the termini to overlap and form a bond. Two stereochemical results are possible. The stereochemical result of the reaction depends on how the two termini rotate. If the termini rotate in the same direction (conrotatory), then the two out groups become trans to each other, the two in groups become trans to each other, and any out group becomes cis to any in group.
On the other hand, if they rotate in opposite directions (disrotatory), then the two out groups become cis to each other, the two in groups become cis to each other, and any out group becomes trans to any in group.
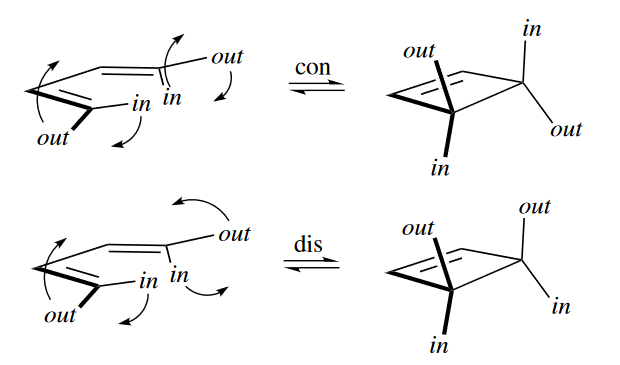
In summary, for butadienes and cyclobutenes: four-electron, thermal, conrotatory; four-electron, photochemical, disrotatory.
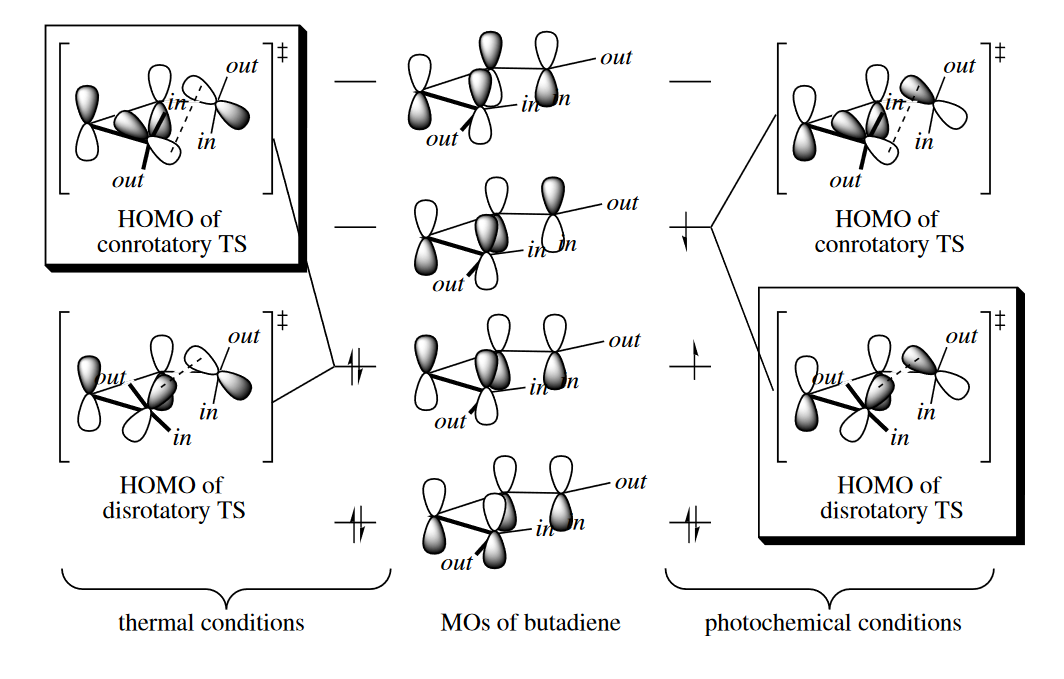
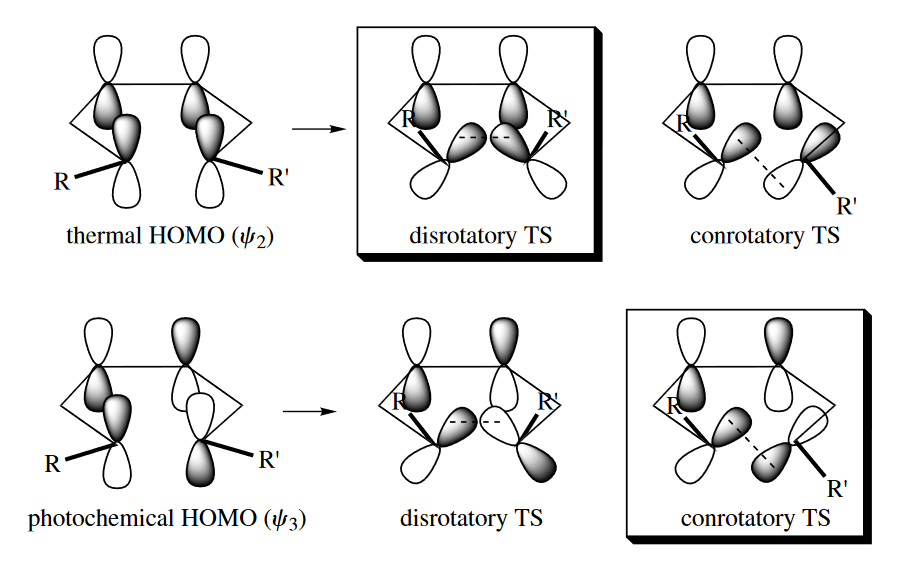
( 这里书上的详细分析我没有多截,根据前线分子轨道理论,电环化关环时形成$\sigma$键需要HOMO轨道的成键作用,所以要看HOMO轨道两端p轨道的对称性来判断顺旋con还是对旋dis。条件不同,HOMO轨道也不同,加热是基态,而光照则处于激发态。从图片应该可以看得比较明白。
至于记忆的话,考虑到轨道的交替对称性,实际上记一个其他就全都得到,不必死记硬背,现想轨道分布实际上也可以。 )
The Woodward–Hoffmann rules for electrocyclic reactions are as follows:
| Woodward-Hoffmann Rules for Electrocyclic Reactions | ||
|---|---|---|
| Number of electron pairs | $\Delta$ | $h\nu$ |
| Odd(2n+1) | disrotatory | conrotatory |
| Even(2n) | conrotatory | disrotatory |
The Woodward–Hoffmann rules for electrocyclic reactions can also be formulated using the terms suprafacial and antarafacial.
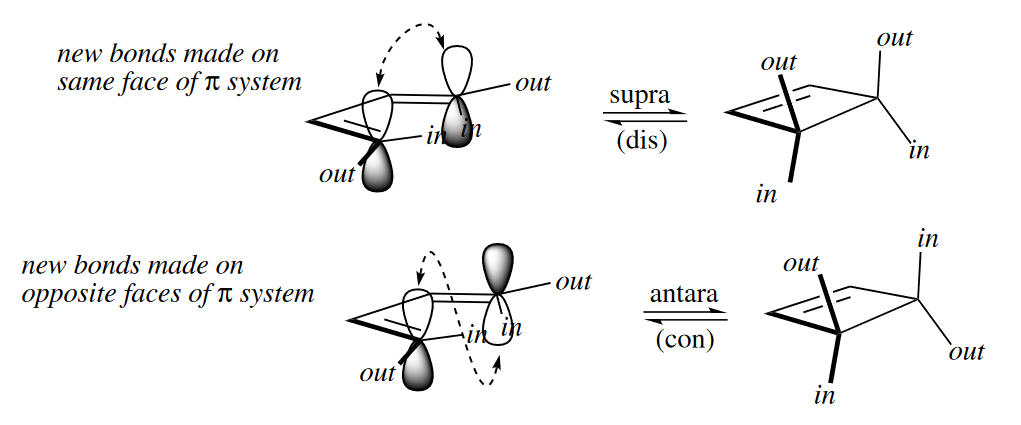
The value of the terms suprafacial and antarafacial is that, unlike disrotatory and conrotatory, they can also be used to describe the way that $\pi$ systems react in cycloadditions and sigmatropic rearrangements. Most importantly, when a $\pi$ system reacts suprafacially, its out groups become cis in the product; when it reacts antarafacially, they become trans.
4.2.3 Stereoselectivity
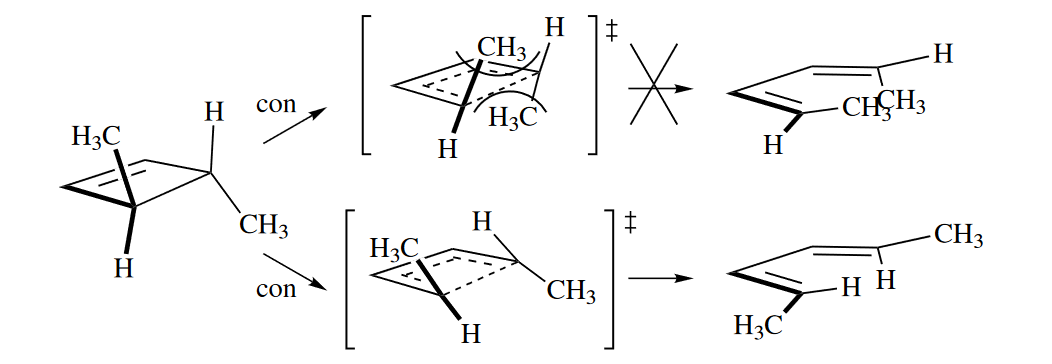
The phenomenon that one “allowed” TS is preferred over the other is called torquoselectivity, a special kind of stereoselectivity. The ring opening of trans-3,4-dimethylcyclobutene is stereospecific because the cis,trans isomer is never obtained, but it is stereoselective because the trans,trans isomer is selectively obtained over the cis,cis isomer.
( 原则上无论是顺旋还是对旋,都有两种转法,但是不是每种转法都能得到对应产物,需要考虑基团位阻的影响,这称为扭转选择性torquoselectivity。由此产生的立体选择性stereoselectivity需要和电环化的立体专一性stereospecific相区分。 )
4.3 Cycloadditions
4.3.1 Typical Reactions
The best-known cycloaddition is the Diels–Alder reaction, a six-electron, [4 + 2] cycloaddition. A 1,3-diene reacts with the $\pi$ bond of a dienophile to give a six-membered ring with one $\pi$ bond. Two new $\sigma$ bonds are formed at the expense of two $\pi$ bonds. The classic Diels–Alder reaction gives a carbocyclic ring, but hetero-Diels–Alder reactions, in which the new ring is heterocyclic, are widely used also.
The Diels–Alder reaction requires that the two double bonds of the diene be coplanar and pointing in the same direction (i.e., in the s-cis conformation). Rotation about the $\sigma$ bond between the internal C atoms of the diene interconverts the s-cis and s-trans conformations. In the s-trans conformation, there are fewer steric interactions between the in groups at the termini of the diene, so dienes normally reside predominantly in this lower energy (by about 4.0 kcal/mol) conformation.
( 环加成对双烯体的构型要求是:必须是s-顺式才能反应,而一般二烯烃的s-反式由于位阻更小,所以更加稳定,也就是说C-C单键的旋转减缓了D-A反应的进行。相应的,如果有环将二烯烃的构型固定为s-顺式,则反应速率加快,若固定为s-反式则无法发生反应。 )
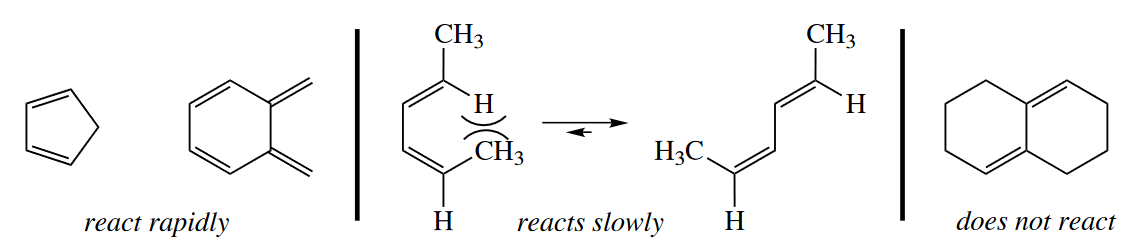
Most Diels–Alder reactions occur with what is called normal electron-demand, in which an electron-rich (nucleophilic) diene reacts with an electron-poor (electrophilic) dienophile. The dienophile may be substituted with carbonyl, $CN$, sulfonyl, $NO_2$, or any other electron-withdrawing group.
As in any reaction, the rate of the Diels–Alder reaction is determined by the energy of its TS. In the TS of most Diels–Alder reactions, the HOMO of the diene interacts with the LUMO of the dienophile.
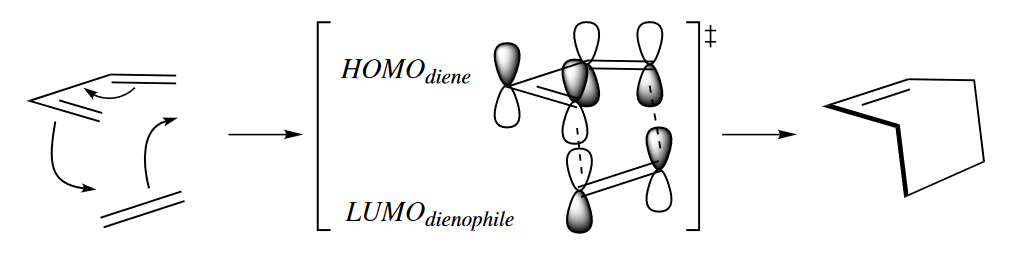
The energy of the Diels–Alder TS is directly related to the strength of the interaction between the MOs, which is in turn related to the difference in energy between the two MOs. The smaller the difference in energy between the two MOs, the stronger their interaction, the lower the energy of the TS, and the faster the reaction.The $HOMO_{diene}$, being a bonding orbital, is lower in energy than the $LUMO_{dienophile}$ . Substitution of the diene with electron-donating groups raises the energy of the $HOMO_{diene}$, bringing it closer to the energy of the $LUMO_{dienophile}$, and substitution of the dienophile with electron-withdrawing groups lowers the energy of the $LUMO_{dienophile}$, bringing it closer to the energy of the $HOMO_{diene}$. Either of these substitutions brings the two orbitals closer in energy, increasing the rate of the reaction.
We mentioned earlier that alkynes are poorer dienophiles than the correspondingly substituted alkenes. The C≡C bond in an alkyne is shorter than the C=C bond in the corresponding alkene, so the C§–C§ overlap is better, and thus the alkyne’s HOMO is lower in energy and its LUMO higher in energy than the alkene’s. The LUMO of the alkyne interacts more poorly with the $HOMO_{diene}$.than the lower energy LUMO of the alkene, so the Diels–Alder reaction of the alkyne is slower.
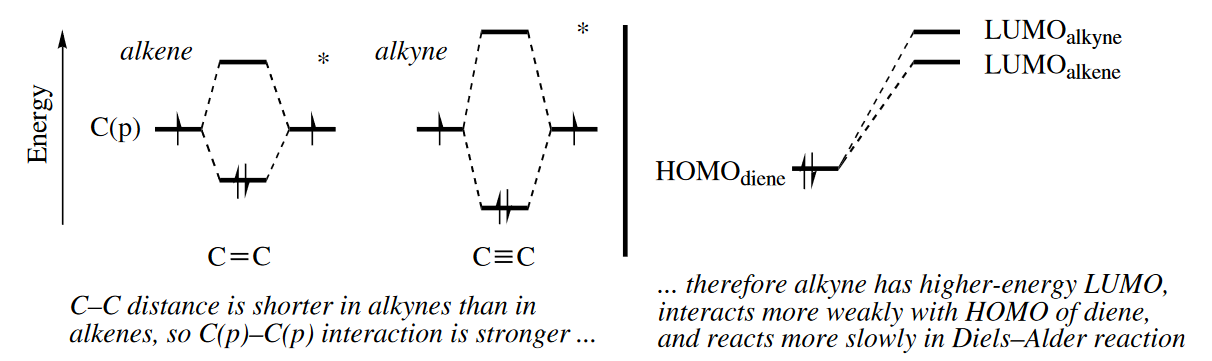
Note that the dependence of the rate of a Diels–Alder reaction on the electronic match between HOMO diene and LUMO dienophile is a kinetic effect, not a thermodynamic one. The reaction of an electron-rich diene with an electron-rich dienophile is just as thermodynamically favorable as its reaction with an electron-poor dienophile, but it does not proceed at a perceptible rate.
( 轨道的能量匹配原则是动力学因素,所以影响的是反应速率。无论是富电子或缺电子的亲双烯体,在反应的热力学趋向上相差不大,但是前者反应速率却极慢,以至于认为不反应。 )
Very electron poor dienes can undergo Diels–Alder reactions with electron-rich dienophiles in the inverse electron-demand Diels–Alder reaction.
Dienes containing heteroatoms such as N and O undergo inverse electron-demand Diels–Alder reactions with electron-rich dienophiles such as enol ethers and enamines. The relatively low energy of the heteroatom p orbitals dramatically lowers the energy of both the $HOMO_{diene}$ and $LUMO_{diene}$.
( 正常的D-A反应是富电子的双烯体和缺电子亲双烯体反应,而缺电子双烯体和富电子亲双烯体反应称为逆电子D-A反应或反常D-A反应。含杂原子的双烯体往往会发生反常D-A反应,因为杂原子p轨道孤对电子的共轭使双烯体的HOMO和LUMO都大幅降低,此时易与富电子亲双烯体反应;如果亲双烯体中含杂原子,如醛或亚胺,那么可以作为缺电子亲双烯体发生正常的D-A反应。 )
1,3-Dipoles react with alkenes and alkynes (dipolarophiles) in 1,3-dipolar cycloadditions (a.k.a. [3 + 2] cycloadditions) to give five-membered heterocycles.
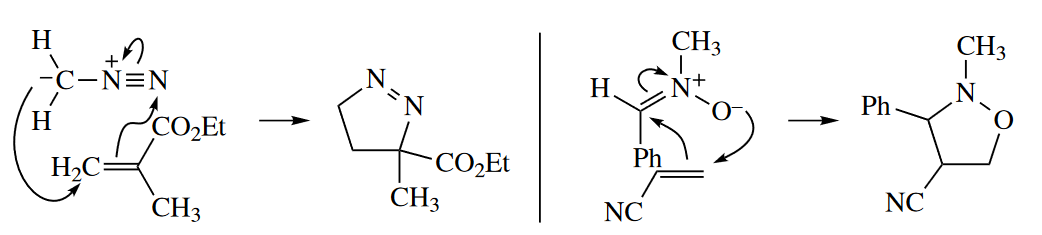
The five-membered heterocyclic product is the key to identifying a 1,3-dipolar cycloaddition. Many 1,3-dipoles are not stable, so they are generated by a series of polar reactions and then react in situ without being isolated.
The selectivity of 1,3-dipoles for electron-rich or electron-poor dipolarophiles is complex. Very electron poor dipoles such as ozone react most quickly with electron-rich and slowly with electron-poor dipolarophiles. Other dipoles, such as azides, react quickly with very electron poor dipolarophiles, slowly with dipolarophiles of intermediate electronic character, and quickly with very electron rich dipolarophiles. This “U-shaped reactivity” is due to a crossover in the nature of the reaction from $LUMO_{dipole} /HOMO_{dipolarophile}$-controlled to $HOMO_{dipole} / LUMO_{dipolarophile}$-controlled as the dipolarophile becomes more electron-poor. At intermediate electronic character, neither the $HOMO_{dipole} /LUMO_{dipolarophile}$ nor the $LUMO_{dipole} /HOMO_{dipolarophile}$ interaction is particularly strong, and the reaction proceeds slowly.
The following are some key ways of identifying cycloaddition reactions:
- All cycloadditions (except cheletropic reactions) form two new $\sigma$ bonds to the termini of two $\pi$ systems.
- If you see a new six-membered ring containing two new bonds with a 1,3-relationship and a $\pi$ bond, think Diels–Alder! A six-membered ring fused to a benzene ring is often made by the [4 + 2] cycloaddition of an o-xylylene and a dienophile or by a [4 + 2] cycloaddition of benzyne and a 1,3-diene.
- If you see a five-membered heterocycle, especially one containing N, think 1,3-dipolar cycloaddition! One of the ring heteroatoms is the central atom of the 1,3-dipole.
- If you see a four-membered ring, think [2 + 2] cycloaddition, especially if the ring is a cyclobutanone (ketene) or light is required (photochemically allowed). Ketenes and other cumulenes undergo [2 + 2] cycloadditions with special facility. An oxetane (four-membered ring with one O) is often obtained from the [2 + 2] photocycloaddition of a carbonyl compound and an alkene.
( 环加成确实是一个好用的成环方法,尤其是在四元环或五元杂环的构建上。如果确定是环加成反应成的环,那么结构其实还是比较容易分析的。 )
4.3.2 Regioselectivity
In fact, because most Diels–Alder reactions proceed by the reaction of nucleophilic dienes with electrophilic dienophiles, the following rule can be formulated as follows: Diels–Alder reactions proceed to put the most electron donating substituent on the diene and the most electron withdrawing substituent on the dienophile either “ortho” or “para” to one another. This “ortho–para rule” misuses the terms ortho and para, which really apply only to benzene rings, but it allows one to remember the regioselectivity of these reactions fairly easily.
Sometimes, the ortho–para rule doesn’t work well, especially when two substituents on the diene have competing directing abilities. For example, a PhS substituent at $C1_{diene}$ is more strongly directing than a MeO substituent at $C2_{diene}$. By resonance arguments, the opposite should be the case, as MeO is a better resonance donor than PhS.
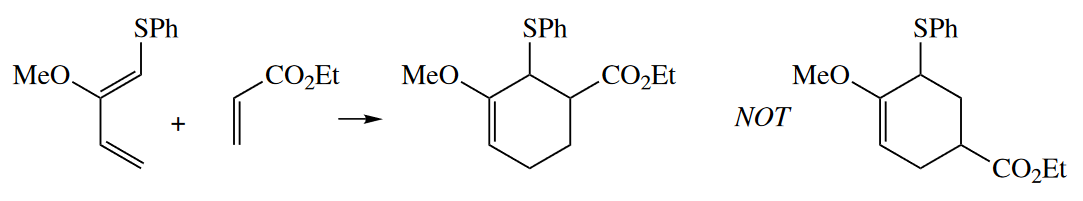
The only way the result can be explained is by looking at the coefficients of the orbitals of the HOMO of the diene and the LUMO of the dienophile.The Diels–Alder reaction proceeds so that the diene terminus with the largest coefficient in the diene HOMO interacts with the dienophile terminus with the largest coefficient in the dienophile LUMO.
( 邻对位选择性对于正常D-A反应还是可以作为判断依据的,但是对于一些特殊反应无法准确判断,而轨道系数的计算需要用到结构化学或量子化学的知识,等学了之后再回头看这个问题吧。 )
4.3.3 Stereospecificity
Some cycloadditions proceed thermally, whereas others require $h\nu$. The dependence of certain cycloadditions on the presence of light can be explained by examining interactions between the MOs of the two reacting components. Frontier MO theory suggests that the rate of cycloadditions is determined by the strength of the interaction of the HOMO of one component with the LUMO of the other.
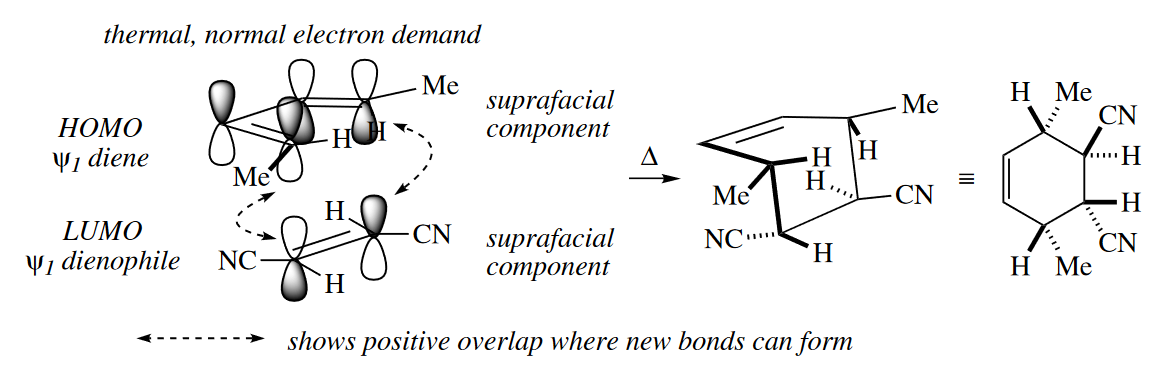
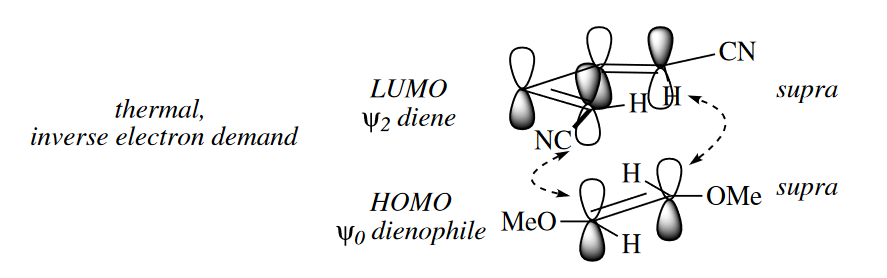
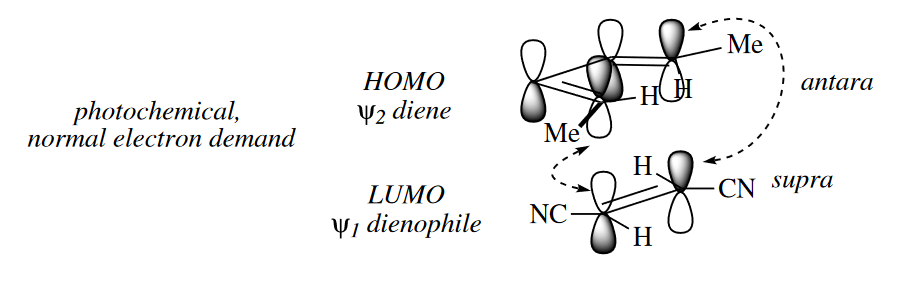
( 这里摘的比较少,略作补充。通过前线分子轨道理论来分析,成键时要求双烯体和亲双烯体的两端轨道的波相一致才能形成成键轨道,如果波相相反则需要先翻转再成键,也就是说要异面成键。通常情况下,异面成键由于位阻和分子几何构型的影响,无法进行,所以只有同面才能顺利进行反应。 )
The stereochemical relationships among substituents in a suprafacial component of a cycloaddition are preserved in the cycloadduct. Groups that are cis(or trans) to one another in the dienophile become cis (or trans) to one another in the product. The two out groups in the diene become cis to one another in the product, as do the two in groups. Because one diastereomeric starting material gives one diastereomeric product, cycloadditions are said to be stereospecific.
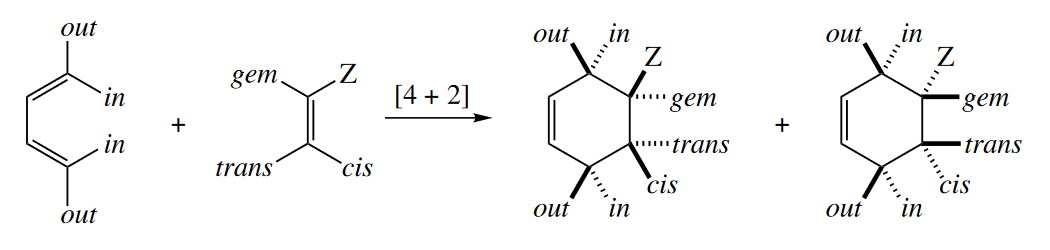
( 在这里有必要理一理立体专一性(Stereospecificity)和立体选择性(Stereoselectivity)的关系。先来看IUPAC给出的定义:
stereospecificity:
A reaction is termed stereospecific if starting materials differing only in their configuration are converted into stereoisomeric products. According to this definition, a stereospecific process is necessarily stereoselective but not all stereoselective processes are stereospecific. Stereospecificity may be total (100%) or partial.
stereoselectivity:
The preferential formation in a chemical reaction of one stereoisomer over another.
总结一下,立体专一性是从构型不同的起始物得到相应的立体异构体,产物的立体构型由原料的立体构型控制;立体选择性是起始物得到两种或多种不同构型的产物,其中一种的产率优于其他产物,这种产率的差异往往是由反应途径导致的。
有说法是立体专一性是立体选择性很高时的表达(“选择”的程度很高所以“专一”),IUPAC说不推荐这种用法,但是实际上许多文献里仍在使用,例如反应产物产量80:20有时会称为80%立体专一性。还有说法认为立体专一性就只有一种特定产物,如$S_N2$的构型翻转,如果出现了多种产物一律认为是立体选择性。两个概念存在很多混用现象,没有确切的答案。
更严谨地区分这两个概念应当从反应机理的角度入手。也就是分析产物的立体构型是来自于反应物立体构型的控制还是反应途径的控制。
)
Under thermal conditions, the TS of the [2 + 2] cycloaddition is made up of $\psi_1$ (the LUMO) of one component and $\psi_0$ (the HOMO) of the other. Positive overlap between the orbitals at both termini of the $\pi$ systems can be obtained only if one of the components reacts antarafacially. This orientation is very difficult to achieve geometrically, and hence [2 + 2] cycloadditions do not normally proceed under thermal conditions. However, under photochemical conditions, one of the components has an electron promoted from $\psi_0$ to $\psi_1$. Now the HOMO–LUMO interaction is between $\psi_1$ of the photoexcited component and $\psi_1$ of the unexcited component, and thus both components can be suprafacial in the TS. The [2 + 2] cycloaddition of most alkenes and carbonyl compounds do in fact proceed only upon irradiation with light.

Three classes of [2 + 2] cycloadditions do proceed under thermal conditions. Ketenes ($R_2C=C=O$) undergo concerted cycloadditions to alkenes under thermal conditions because the ketene can react antarafacially with an alkene that reacts suprafacially.
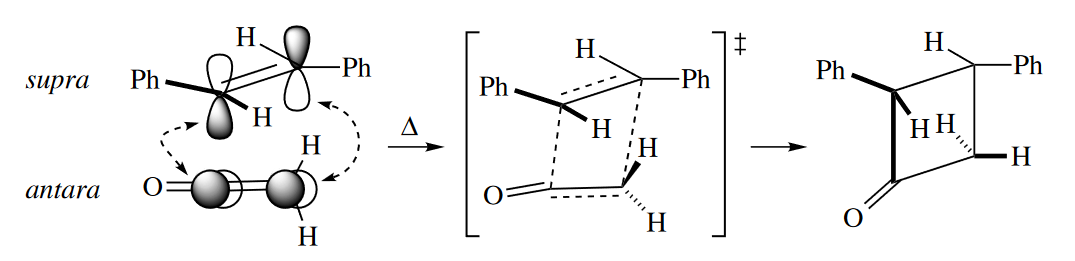
( 烯酮可以“侧过来”发生轨道的重叠,主要原因是因为羰基碳原子周围的位阻很小。而对于一般烯烃,如果也像这样侧过来实现异面轨道的重叠,那么碳上连接的基团(氢原子或其他基团)的位阻会导致排斥力太大,重叠效果差,所以一般烯烃是不能在加热条件发生类似的[2+2]环加成的。 )
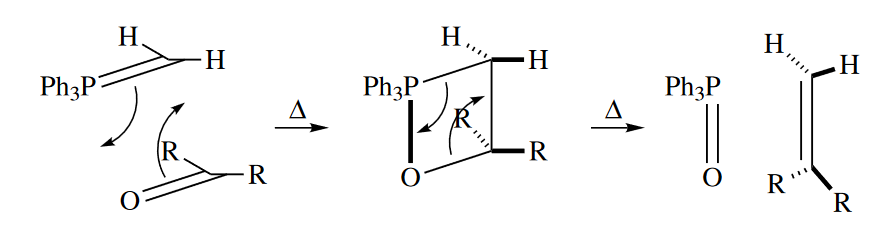
The second kind of thermally allowed [2 + 2] cycloaddition occurs when one of the atoms involved is a second-row or heavier element, as in the Wittig reaction.Why aren’t the [2 + 2] cycloaddition and retro-cycloaddition of the Wittig reaction disallowed? The Wittig reaction proceeds at ordinary temperatures because when $\pi$ bonds to elements heavier than C, N, or O are involved in pericyclic reactions, factors other than mismatches in MO symmetries become much more important to the energy of the TS. In other words, the concerted mechanism for the Wittig reaction is disallowed by the Woodward–Hoffmann symmetry rules, but it is not as disallowed as the [2 + 2] cycloadditions of $C=C$ or $C=O$ $\pi$ bonds. Other factors, including the poor overlap of the orbitals comprising the $C=P$ $\pi$ bond, the very high energy of that bond, and the very low energy of the $P-O$ $\sigma$ bond, lower the energy of the TS of the Wittig reaction enough that it proceeds at ordinary temperatures.
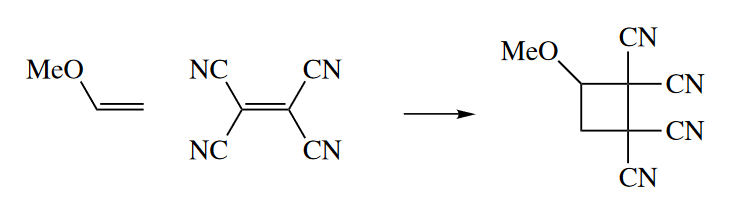
In the third class of thermally allowed [2 + 2] cycloadditions, a very electron rich alkene is allowed to react with a very electron poor diene. These reactions almost certainly proceed stepwise through a zwitterionic or diradical intermediate, so they are not pericyclic reactions, and no violation of the Woodward–Hoffmann rules occurs.
To summarize, most [2 + 2] cycloadditions are light-promoted. The only concerted thermal [2 + 2] cycloadditions involve a ketene or other cumulene, or a compound in which a heavy atom such as P or a metal is doubly bound to another element.
The Woodward–Hoffmann rules for cycloadditions are as follows:
| Woodward–Hoffmann Rules for Cycloadditions | ||
|---|---|---|
| Number of electron pairs | $\Delta$ | $h\nu$ |
| Odd | suprafacial–suprafacial | suprafacial–antarafacial |
| Even | suprafacial–antarafacial | suprafacial–suprafacial |
( 由于位阻的影响,通常只有s-s的构型才能顺利发生环加成,s-a的反应通常很难进行。 )
The application of the Woodward–Hoffmann rules to cheletropic reactions is not straightforward. Some results are difficult to rationalize or predict a priority.
4.3.4 Stereoselectivity
Diels–Alder reactions generally proceed selectively via the TS in which the most powerful electron-withdrawing group on the dienophile is endo, i.e., sitting underneath the diene, as opposed to pointing away from it. This phenomenon is known as the endo rule.
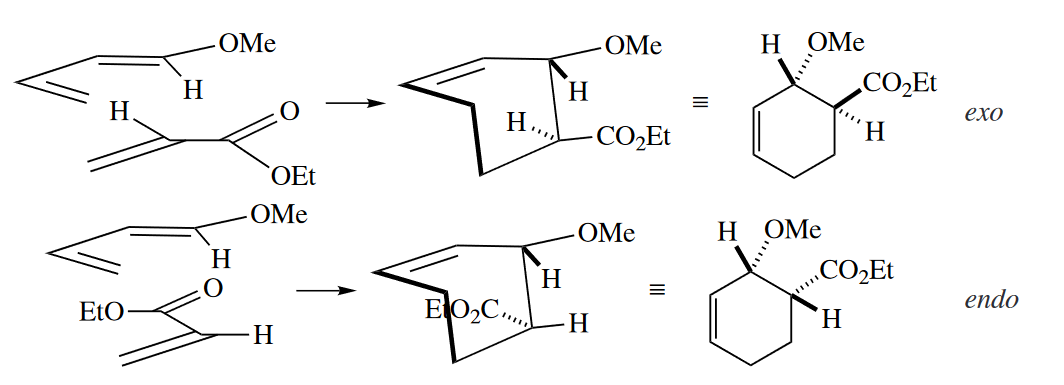
The out-endo-cis rule is a device for drawing the products of Diels–Alder reactions with stereochemistry consistent with the endo rule. The first word refers to the orientation of a group on a terminus of the diene. The second word refers to a substituent on the dienophile (usually the most electron withdrawing one). The third word gives the stereochemical relationship of these two groups in the product. Thus, the out group on the diene and the endo group on the dienophile are cis in the product.
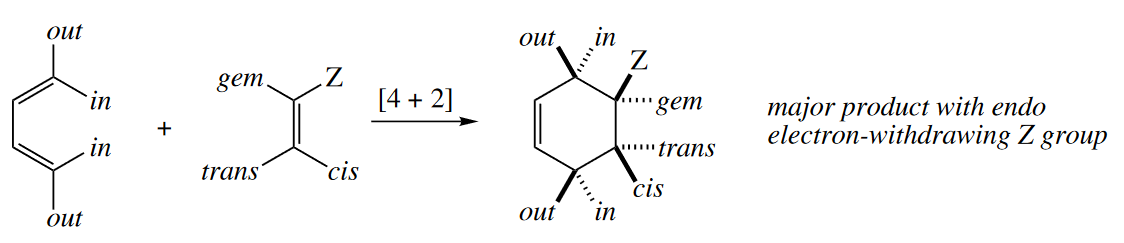
The endo rule applies equally to inverse electron-demand Diels–Alder reactions. In these reactions, the most electron donating group on the dienophile is preferentially endo. The out-endo-cis rule applies, too.
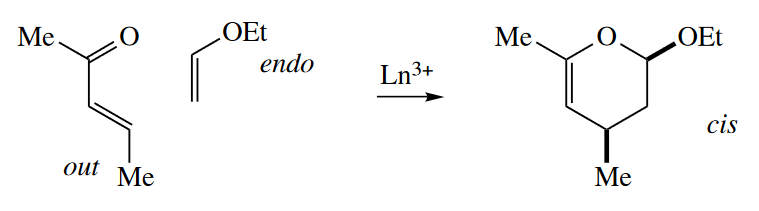
( out-endo-cis规则对于正常的D-A反应和反常的D-A反应都适用,但是两种反应中endo group的选择是不同的,正常D-A反应中endo group是亲双烯体中吸电子能力最强的基团,而反常D-A反应则是亲双烯体中给电子能力最强的基团。 )
Why is the more sterically encumbered TS lower in energy? The most widely accepted explanation cites secondary orbital interactions. In the more crowded approach, the orbitals of the carbonyl group of the dienophile can interact with the orbital on C2 of the diene. The secondary orbital interactions in the endo TS are energetically favorable, so the kinetic product is the more crowded, less thermodynamically stable endo product.
Endo selectivity is a kinetic phenomenon. If an equilibrium is established between the higher energy endo and lower energy exo products, then predominantly exo products will be seen.
1,3-Dipolar cycloadditions give predominantly endo products, too. Again, the out-endo-cis rule applies.
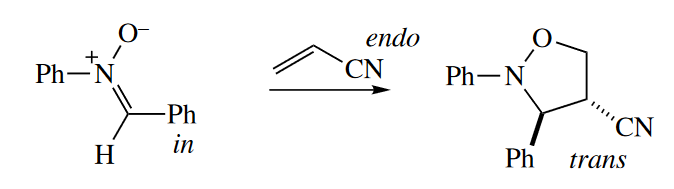
[2 + 2] Photocycloadditions also proceed to give predominantly endo products.
In the [2 + 2] cycloaddition of monosubstituted ketenes to cycloalkenes, two products can also be obtained. The thermodynamic product, where the R group is on the convex face of the bicyclic system, is not obtained predominantly. In fact, the reaction is “masochistic”: the larger the R group, the greater the proportion of the thermodynamically less stable product.
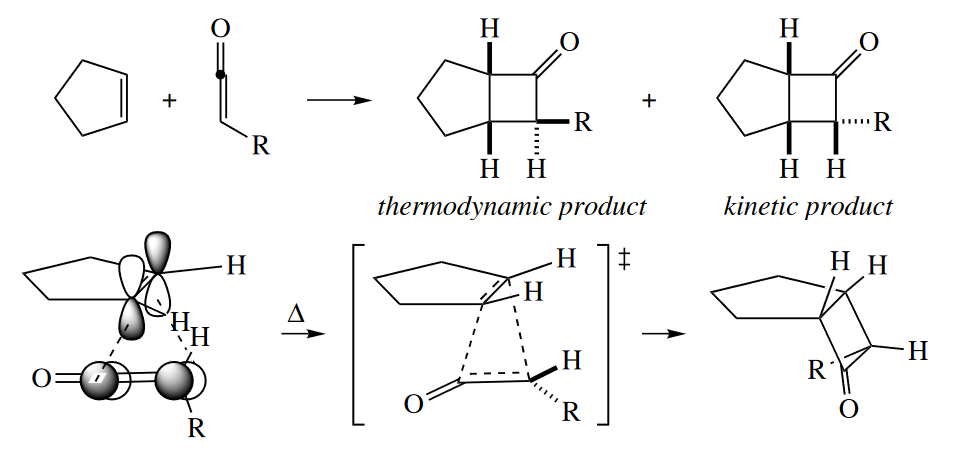
( R基团越大,则动力学产物越多。烯酮环加成的这种反常现象仍然是因为它反常的异面加成机理,在发生异面加成时,烯酮发生扭转,R基团被扭向下方,此时TS的能量小,所以反应在动力学上有利,从而得到动力学产物。 )
4.4 Sigmatropic Rearrangements
4.4.1 Typical Reactions
The [3,3] sigmatropic rearrangements (Cope and Claisen rearrangements) are the most widely used sigmatropic rearrangements and are probably the most widely used pericyclic reactions after the Diels–Alder reaction. In the Cope rearrangement, a 1,5-diene isomerizes to another 1,5-diene. In the Claisen rearrangement, an allyl vinyl ether (a 1,5-diene in which one C atom is replaced with O) isomerizes to a $\gamma$,$\delta$-unsaturated carbonyl compound (another 1,5-diene in which one C atom is replaced with O).
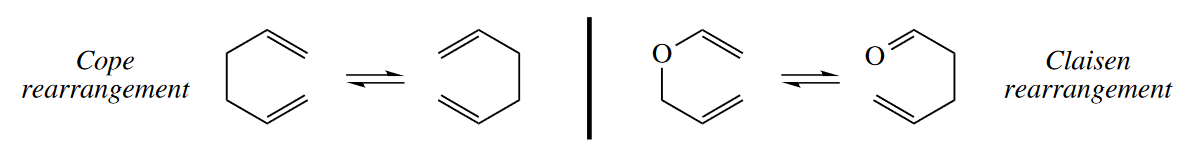
he key to identifying Cope and Claisen rearrangements is the 1,5-diene in the starting material or in the product. A $\gamma$,$\delta$-unsaturated carbonyl compound (a 1,5-heterodiene) can be made by a Claisen rearrangement, and a $\delta$,$\epsilon$-unsaturated carbonyl compound can be made by an oxy-Cope rearrangement.
( 氧杂Cope重排和Claisen重排的差别可以从重排的骨架分辨,如果在[3,3]$\sigma$重排中参与标号的六个原子有氧原子,则是Claisen重排,反之为氧杂Cope重排。 )
The [1,5] sigmatropic rearrangement of H atoms around a cyclopentadienyl group is an extremely facile process because the ends of the pentadienyl system are held closely to one another. The different isomers of a substituted cyclopentadiene are in rapid equilibrium (on the laboratory time scale) above 0 °C.
The [1,5] alkyl shift is also seen in cyclopentadienes, but much higher temperatures (usually >200 °C) are required. The C($sp^3$) orbital is much more directional than the H(s) orbital, so the simultaneous overlap with two orbitals that is required in the TS is not as favorable as it is with H.
The [2,3] sigmatropic rearrangement involves the rearrangement of a 1,2-dipole to a neutral compound. An allyl group migrates from the positive end to the negative end of the dipole to neutralize the charges.Amine oxides, sulfoxides, and selenoxides all undergo the [2,3] sigmatropic rearrangement.
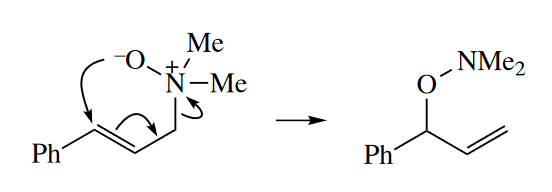
The [2,3] sigmatropic rearrangement of alloxycarbanions is known as the Wittig rearrangement (not to be confused with the Wittig reaction). The requisite carbanions can be prepared by transmetallation of a stannane (tin compound). Allyl propargyl ethers are also good substrates for the Wittig rearrangement. Deprotonation of the propargyl position is relatively facile.
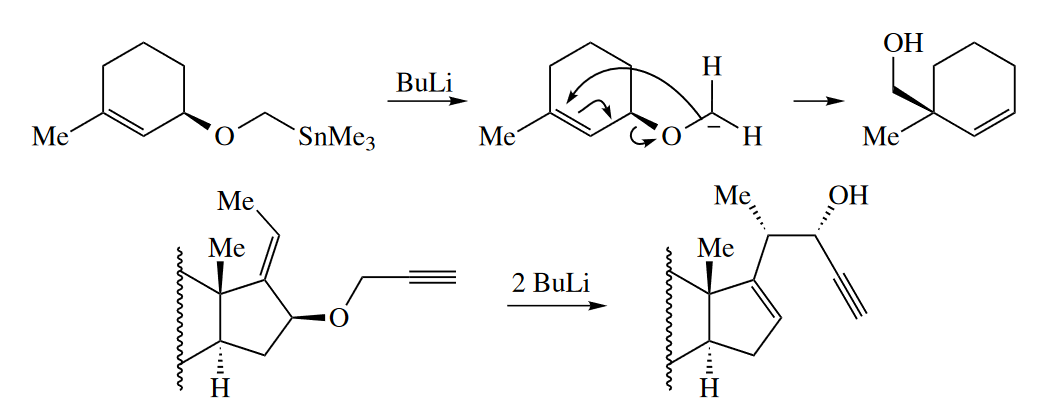
Four-electron, [1,3] sigmatropic rearrangements are very rare. Occasionally an alkyl group can undergo a concerted 1,3-shift, but H atoms never undergo concerted [1,3] sigmatropic rearrangements.
( [1,3]迁移是异面迁移,所以氢原子不能发生,详细解释在下一节。另外,虽然烯醇互变看起来很像是“H的[1,3]迁移”,但是烯醇互变不是协同反应,它是在酸碱催化下进行的极性机理。 )
The [1,7] sigmatropic rearrangement, an eight-electron process, is also quite rare. A very important [1,7] sigmatropic rearrangement, though, occurs
in the human body. Precalciferol (provitamin D 2) is converted to ergocalciferol (vitamin D 2) by a [1,7] sigmatropic H shift.
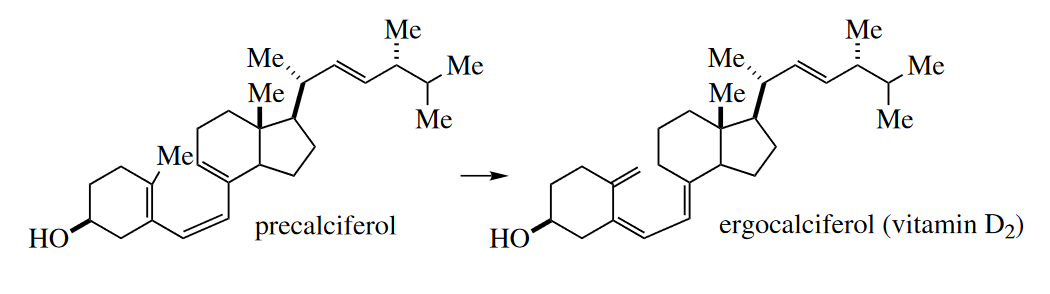
All the sigmatropic rearrangements discussed so far occur under thermal conditions. Photochemical sigmatropic rearrangements are extremely rare.
4.4.2 Stereospecificity
The most common sigmatropic rearrangements are [1,2] cationic, [1,5], and [3,3] rearrangements. These rearrangements proceed readily under thermal conditions.
The H atom is always classified as a suprafacial component, as the 1s orbital is monophasic, so positive overlap in the TS between orbitals where bond-making and bond-breaking take place is produced when the two-atom component is suprafacial.
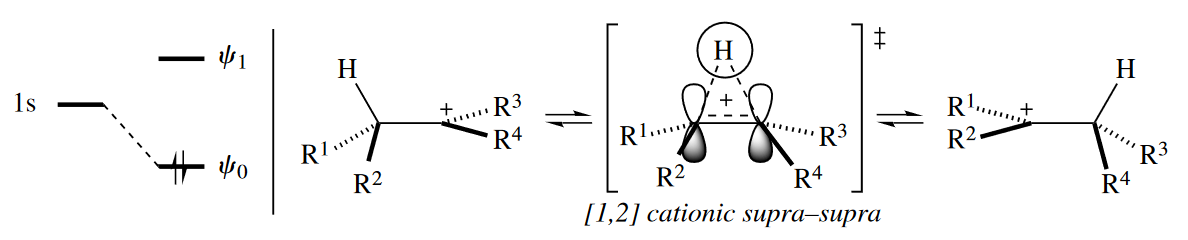
Likewise, in the cationic [1,2] alkyl shift, both components must be suprafacial for there to be positive overlap in the TS between orbitals where bond-making and bond-breaking take place. The migrating group retains its configuration because of the requirement for suprafaciality.
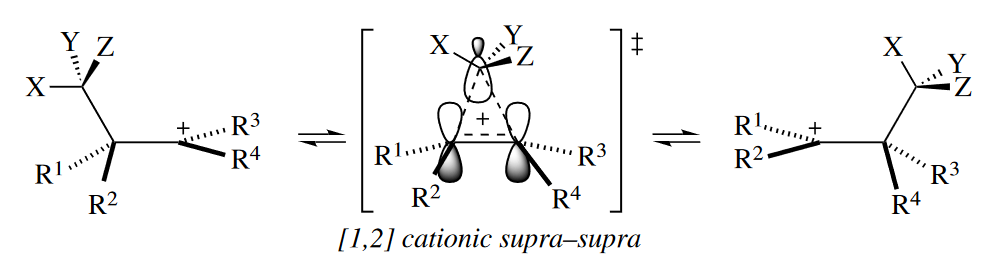
The dominant FMO interaction in the TS of the [1,5] sigmatropic rearrangement, a six-electron reaction, is between the H(1s) orbital and $\psi_2$ of the five-atom component, no matter how the six electrons are divided between the two components. (An interaction between two half-filled orbitals is shown). Again, positive overlap in the TS between orbitals where bond-making and bond-breaking take place is produced when the five-atom component reacts suprafacially.
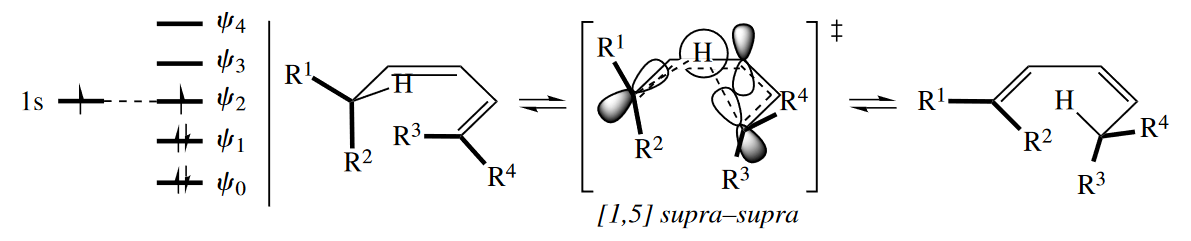
The [3,3] sigmatropic rearrangement is a six-electron reaction. No matter how the six electrons are distributed between the two three-atom components, the dominant FMO interaction in the TS is between $\psi_1$ of one component and $\psi_1$ of the other component. The reaction proceeds suprafacially with respect to both components.
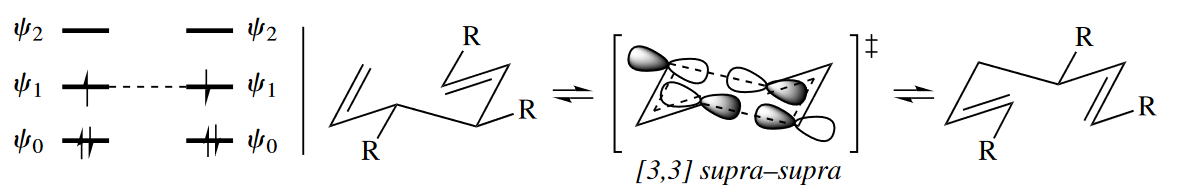
By contrast, in the [1,2] anionic H shift, the dominant FMO interaction is between the 1s orbital of the one-atom component and $\psi_1$ of the two-atom component. For there to be positive overlap between orbitals where bond-making and bond-breaking take place, the two-atom component must react antarafacially. Because
this arrangement is geometrically impossible, [1,2] anionic H shifts are thermally disallowed reactions.
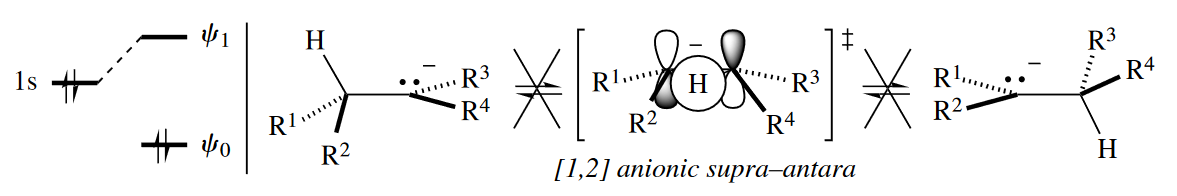
A thermally allowed anionic [1,2] shift can occur if the one-atom component reacts antarafacially. In the case of the alkyl shift, the C§ orbital of the migrating R group can react antarafacially, with the configuration inverting upon migration. In fact, the geometric requirements for anionic [1,2] alkyl shifts are so stringent that the reactions are extremely rare.
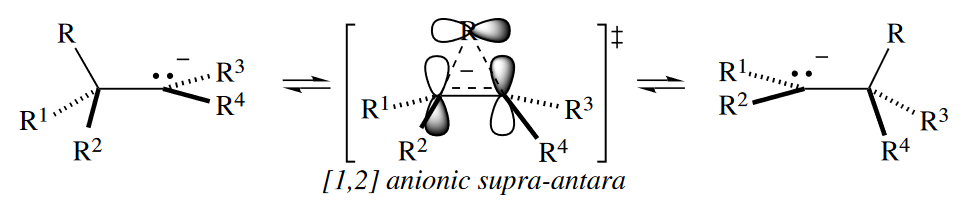
( 这里电子数的不同导致了参与反应的分子轨道不同,所以轨道对称性影响了反应结果。对于氢原子来说,其1s轨道球形对称,只能与另一部分同面迁移,而不能异面迁移;对于烷基来说,C的2p轨道具有两个波相,所以可以翻转构型来实现异面迁移。但是注意,即使是后者,类似的反应也非常少见,因为位阻的影响仍然很大。 )
Thermal [1,3] H shifts such as the concerted rearrangement of enols to carbonyl compounds are disallowed. The Woodward–Hoffmann rules, though, say that one of the two components of this four-electron rearrangement must react antarafacially for it to be allowed. Therefore this rearrangement reaction always proceeds through a nonconcerted mechanism and requires acidic or basic catalysis.
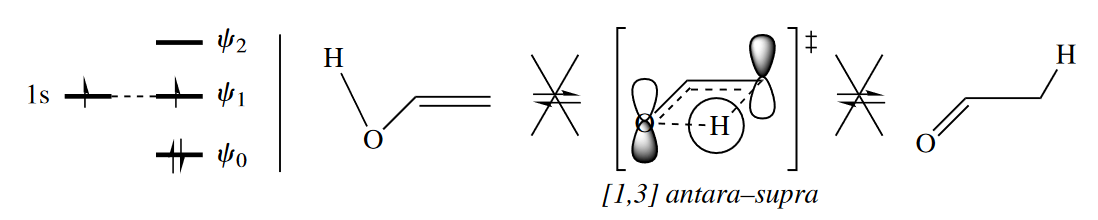
Thermal [1,3] sigmatropic rearrangement is allowed only if one component is antarafacial, but a photochemical [1,3] sigmatropic rearrangement is expected to be allowed when it proceeds suprafacially with respect to both components. The stereochemical requirement changes because under photochemical conditions, the HOMO of the three-atom component is $\psi_2$ (symmetric), not $\psi_1$ (antisymmetric).
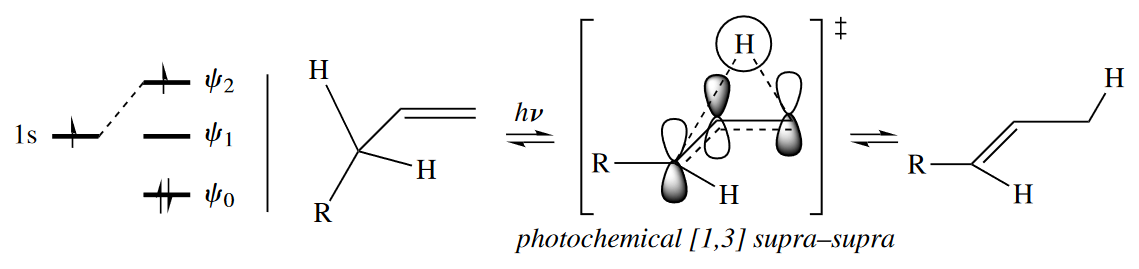
4.4.3 Stereoselectivity
Cope rearrangements are required to be suprafacial with respect to both components, but multiple stereochemical results are still possible. Cope rearrangements have a six-membered ring in the TS. The ring can be in one of four conformations (two chair and two boat). These different conformers can lead to different stereoisomeric products.
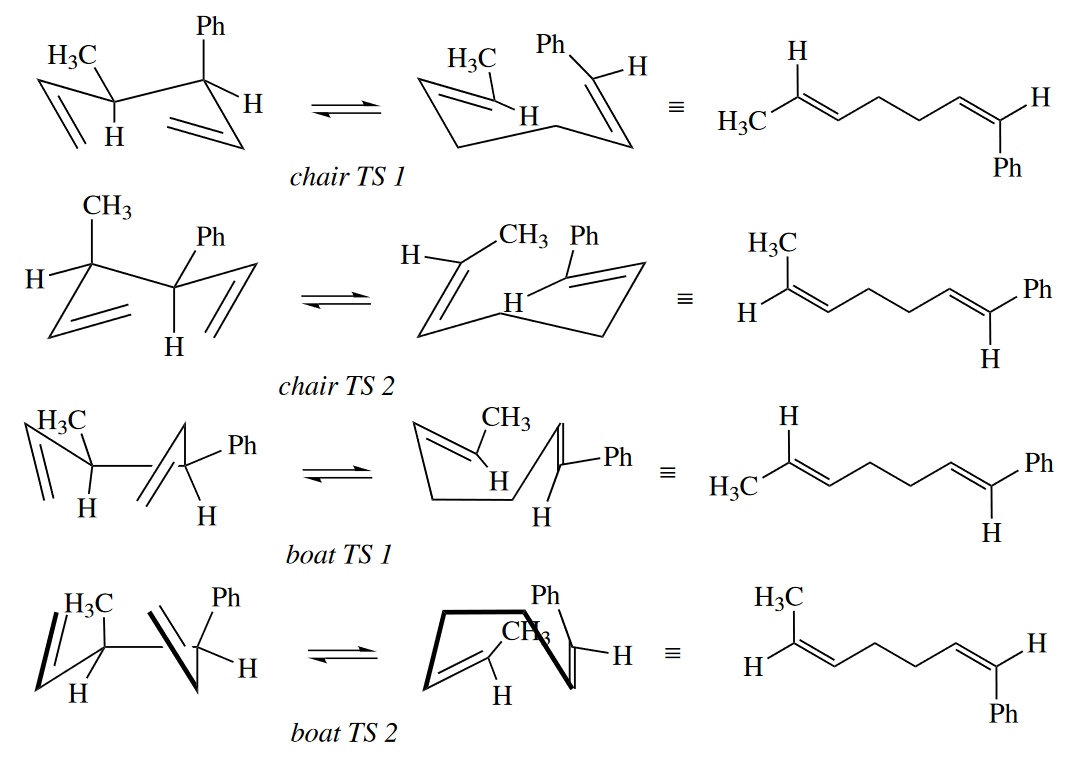
Of course, one of the four possible TSs is usually quite a bit lower in energy than the others, and this leads to stereoselectivity in the Cope rearrangement. The chair TSs are preferred to the boat TSs, just as they are in cyclohexanes.
( 通常来说椅式构象较船式构象更稳定,所以一般的Cope/Claisen重排经历椅式中间体,并且选择能量最低,最稳定的椅式中间体来得到反应产物,其考虑的因素主要是取代基位阻的影响,如大位阻基团一般占据平伏键位置。 )
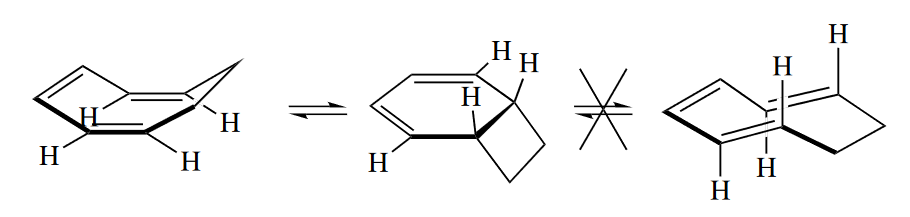
Not every Cope or Claisen rearrangement can proceed through a chair TS. Sometimes the chair TS is raised prohibitively high in energy compared with the boat TS. For example, cis-1,2-divinylcyclopropane undergoes a Cope rearrangement to give 1,4-cycloheptadiene. If the starting material underwent rearrangement through the chair TS, one of the $\pi$ bonds in the seven-membered ring in the product would be trans, so the reaction proceeds through the boat TS.
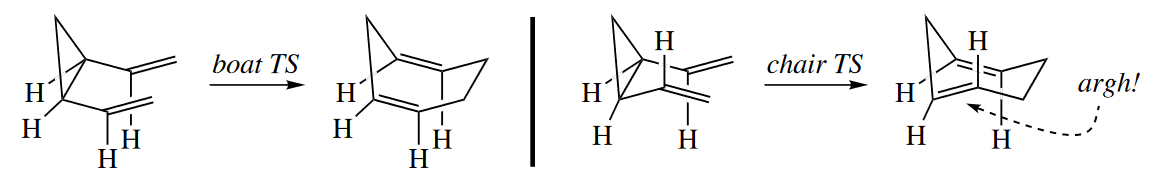
For any 1,5-diene other than a simple, acyclic one, molecular models are usually necessary to determine whether the Cope or Claisen rearrangement will proceed through a chair or a boat TS.
( 但是如果椅式构象能量太高,如形成了环内E型双键等,则会经历船式构象。 )
Cope and Claisen rearrangements are not the only sigmatropic rearrangements that show stereoselectivity. For example, consider the [1,5] sigmatropic rearrangement of the following diene.
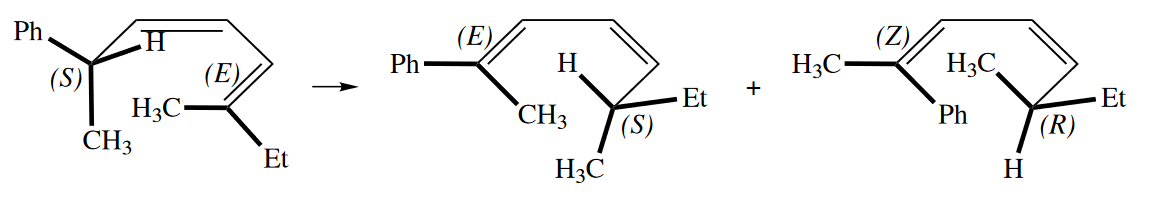
A similar argument can be made for the [1,7] sigmatropic rearrangement.
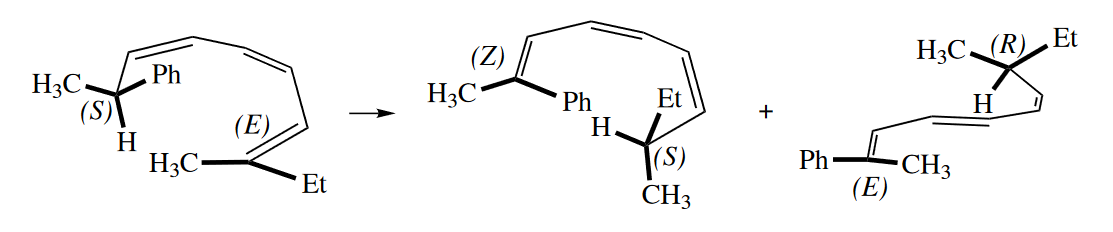
( [1,5]H迁移是同面迁移,双键构型都不变;[1,7]H迁移是异面迁移,所以会有一个双键扭转的产物。 )
4.5 Ene Reactions
The ene reaction is always a six-electron reaction. Like the Diels–Alder reaction, it has one four-electron component, the ene, and one two-electron component, the enophile. The two-electron component is a $\pi$ bond. The four-electron component consists of a $\pi$ bond and an allylic $\sigma$ bond. The atom at the terminus of the $\sigma$ bond is usually H; the other five atoms involved in the ene reaction may be C or heteroatoms. Because the ene reaction involves six electrons, it is suprafacial with respect to all components.
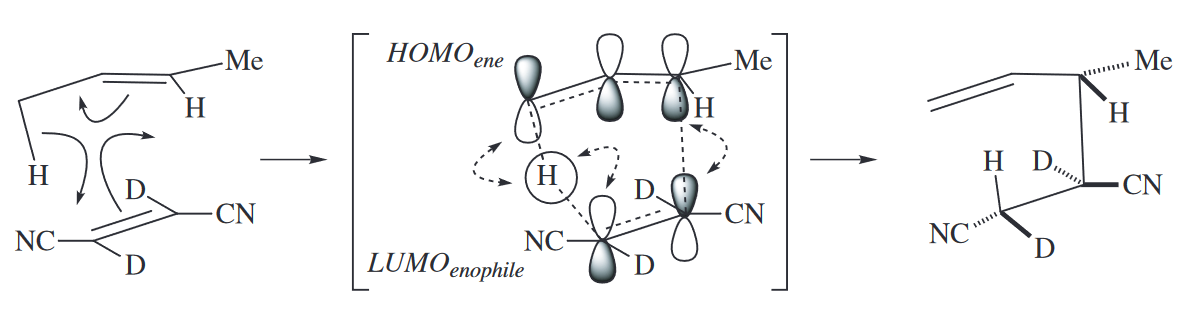
Ene reactions involving five C atoms and one H usually require very high temperatures (> 200 °C) to proceed. The reaction occurs at much lower temperature if the H atom is replaced with a metal atom in the metalla-ene reaction. The metal may be Mg, Pd, or another metal.
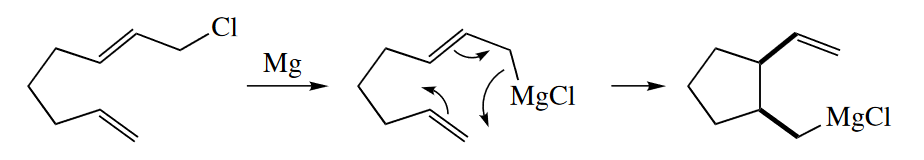
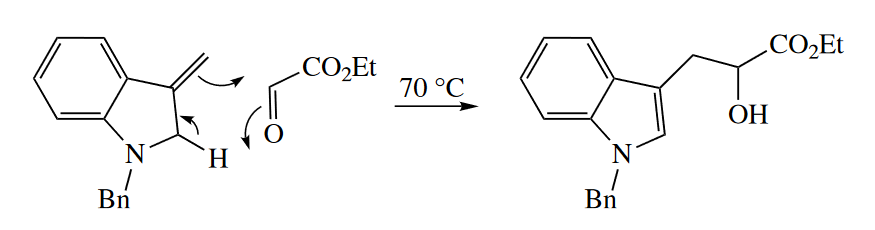
The ene reaction also occurs when heteroatoms replace C in either the ene or the enophile. The following hetero-ene reaction requires a much lower activation energy than normal because of the driving force of formation of the aromatic indole ring and because of the instability of ethyl glyoxylate, which has adjacent carbonyl groups.
A number of synthetically useful elimination reactions proceed thermally, with no base or acid required. These elimination reactions proceed through a concerted retro-ene mechanism. (The mechanism is sometimes called $E_i$.) The thermal elimination of acetic acid from alkyl acetates and the elimination of $RSCOSH$ from alkyl xanthates (the Chugaev reaction) are retro-ene reactions.
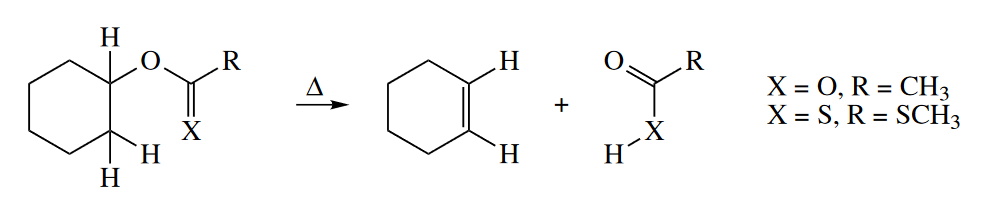
Retro-ene reactions are partly driven by the gain in entropy. (Entropic contributions to $\Delta G^{\ominus}$ are much more important at the high temperatures required for ene reactions.) The formation of the $C=O$ $\pi$ bond at the expense of the $C=S$ $\pi$ bond provides an additional driving force for the Chugaev reaction. Acetic acid elimination is enthalpically unfavorable, and it requires much higher temperatures so that the entropic contribution becomes dominant. The formation of the $C=O$ $\pi$ bond and entropic gains also provide the driving force for the elimination of methyl formate from the following allylic MOM (methoxymethyl) ether.
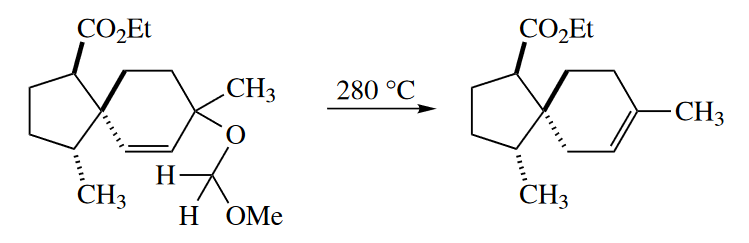
( 普通酯的热裂焓效应上是不利的,所以需要更高的温度,此时熵效应具有主要作用;而黄原酸酯的热裂(Chugaev反应)中$C=S$双键转化为$C=O$双键是一个有力因素,因此温度需求更低。 )
Ene reactions make one new $\sigma$ bond at the expense of one $\pi$ bond, like electrocyclic reactions, but they are fairly easy to distinguish from electrocyclic reactions otherwise. Look for allylic transposition of a double bond and transfer of an allylic H atom. In a retro-ene reaction, a nonallylic H is transferred to an allylic position.
4.6 Summary
( 摘取部分内容翻译。 )
General considerations when drawing pericyclic mechanisms:
- In many problems, a series of polar reactions proceeds to give a reactive intermediate that then undergoes a pericyclic reaction to give the product. Look at the product, and determine what pericyclic reaction could generate it. Then draw the immediate precursor to the final product. Don’t be shy about drawing curved arrows on the product to help you draw the precursor with all its bonds in the correct position. This procedure often simplifies the problem considerably. 在许多问题中,一系列极性反应给出的中间体再经过周环反应生成产物。观察产物,并确定是由哪种周环反应生成的。接着画出最终产物的前体。不要不敢去画箭头,这将帮助你画出成键关系正确的前体。这些步骤往往能有效简化问题。
- As always, draw your by-products, label your atoms, make a list of bonds to make and break, and obey Grossman’s rule! 总是要写出副产物,给原子标号,列出成键和断键的列表,以及遵守Grossman规则(笑)。
Whether working forward or backward, look for some key substructures.
- The presence of a 1,3-butadiene or a cyclobutene in the starting material or the product may indicate a four-electron electrocyclic reaction. 反应物或产物中有1,3-丁二烯或环丁烯预示着4e电环化。
- The presence of a 1,3-cyclohexadiene or a 1,3,5-hexatriene in the starting material or the product may indicate a six-electron electrocyclic reaction. 反应物或产物中有1,3-环己二烯或1,3,5-己三烯预示着6e电环化。
- Cyclopropyl cations and allylic cations can open and close, respectively, by two-electron electrocyclic reactions, as can the corresponding halides. 环丙基正离子/烯丙基正离子可以分别通过2e电环化开环/关环,相应的卤化物也可以。
- A six-membered ring with two new $\sigma$ bonds in a 1,3-relationship may indicate a [4 + 2] cycloaddition (Diels–Alder or hetero-Diels–Alder reaction). When the new ring is fused to an aromatic one, benzyne or an o-xylylene may have been a reactive intermediate. 六元环中有两根新形成的$\sigma$键预示着[4 + 2]环加成(D-A或杂原子D-A)。当新形成的环并一个芳环,那么苯炔或邻苯碳醌(邻苯醌的氧被碳取代)可能是反应中间体。
- The formation of a five-membered heterocycle with two new $\sigma$ bonds almost always indicates a [3 + 2] cycloaddition. 形成两根新$\sigma$键并成五元杂环预示着[3 + 2]环加成。
- Light and a cyclobutane often indicate a [2 + 2] cycloaddition. 光照条件和环丁烷通常预示着[2 + 2]环加成。
- Loss of $CO_2$, $N_2$, $CO$, or $SO_2$ by cleavage of two $\sigma$ bonds often indicates a retro [4 + 2] or [4 + 1] cycloaddition. 断开两根$\sigma$键离去一分子$CO_2/N_2/CO/SO_2$通常预示着逆[4 + 2]/逆[4 + 1]环加成。
- A 1,5-diene, including a $\gamma$,$\delta$-unsaturated carbonyl compound, often indicates a [3,3] sigmatropic rearrangement, i.e., a Cope or Claisen rearrangement. 1,5-二烯通常预示着[3,3]-$\sigma$重排,例如Cope/Claisen重排。
- The H atoms of cyclopentadienes undergo [1,5] sigmatropic rearrangements with great facility. 环戊二烯的H原子很容易发生[1,5]-H迁移。
- Migration of an allylic group from a heteroatom to its next-door neighbor often indicates a [2,3] sigmatropic rearrangement. 烯丙基从杂原子向相邻原子迁移预示着[2,3]-$\sigma$重排。
- The formation of a new $\sigma$ bond at the ends of two $\pi$ bonds and the simultaneous migration of a H atom may indicate an ene reaction. 在两个$\pi$键末端生成新$\sigma$键,同时伴随着H迁移,预示着ene反应。
- The elimination of an acid such as AcOH or PhSeOH may indicate a retro-ene or retro-hetero-ene reaction. 羧酸(如乙酸或硒酸)的消除可能预示着逆ene反应或逆杂原子ene反应。
- A requirement of light ($h\nu$) suggests that either an electrocyclic reaction (usually a ring closing), an even-electron-pair cycloaddition (e.g., a [2 + 2] cycloaddition), or a free-radical reaction has occurred. 反应需要光照说明可能发生了电环化反应(通常是关环)、偶数对电子的环加成(4n e,如[2 + 2])或自由基反应。
Good luck!
第四章是周环反应,翻完这一章之后对周环反应的理解就可以说是比较全面了,从前线分子轨道入手来讲解了周环反应的四大类型,并且配以大量的例子和解释,也涉及了周环反应的立体化学内容,过一遍可以巩固不少常见的反应套路。
周环反应的例子都还挺熟悉的,不过要想周密地写出陌生反应机理还是需要依靠大量练习来掌握反应条件和反应的立体化学。Woodward-Hoffman规则不必死记硬背,通过对称性交替变化的规律只需要记住一个例子就能自然推出其他规律。
习题之后再补…
Chapter Ⅳ
2. Predict the major product (regio- and stereoisomer) of each of the following cycloadditions. All reactions but (h) are [4 + 2] or [3 + 2] cycloadditions.
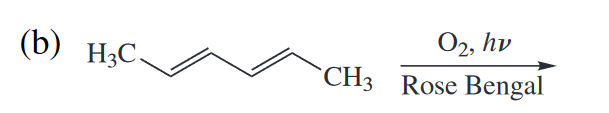
Ans
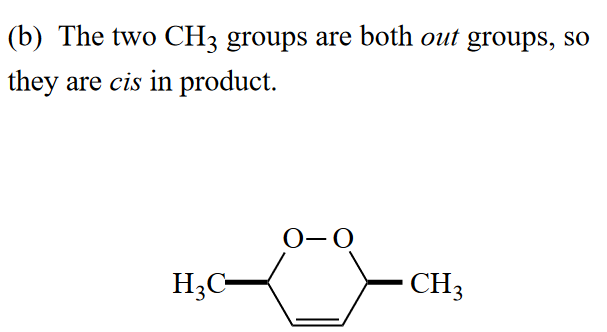
此处的氧气作在光照激发下和双烯体发生[4 + 2]环加成。
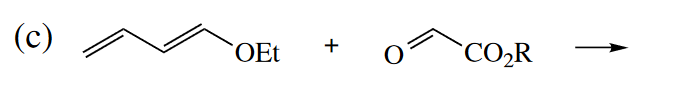
Ans
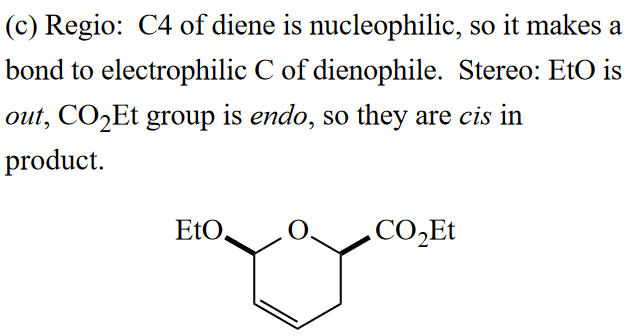
本题亲双烯体为碳氧双键,所以羰基碳是亲电位点,以此来确定区域选择性。
立体选择性依然满足out-endo-cis规则。
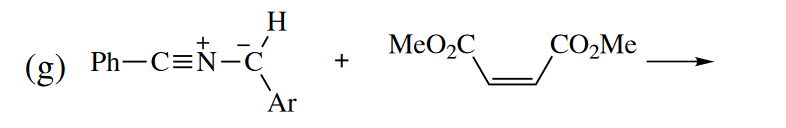
Ans
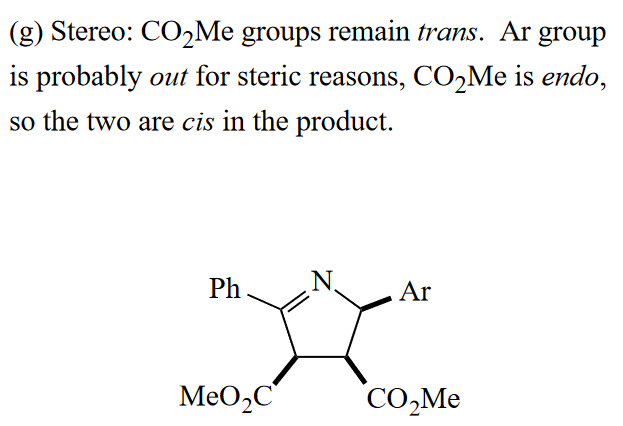
本题需要判断芳基Ar的位置,考虑位阻的影响,所以芳基基团可能是处于out group的位置,那么根据out-endo-cis规则,可得到最后的构型。
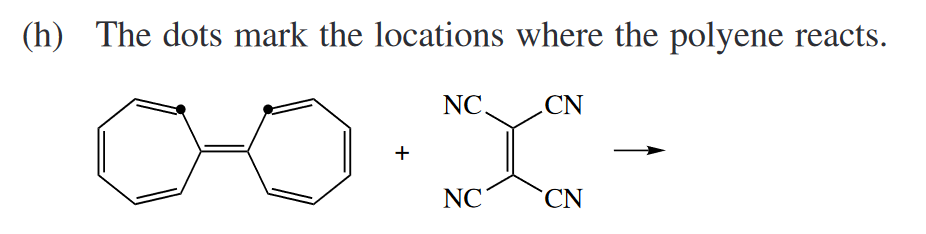
Ans
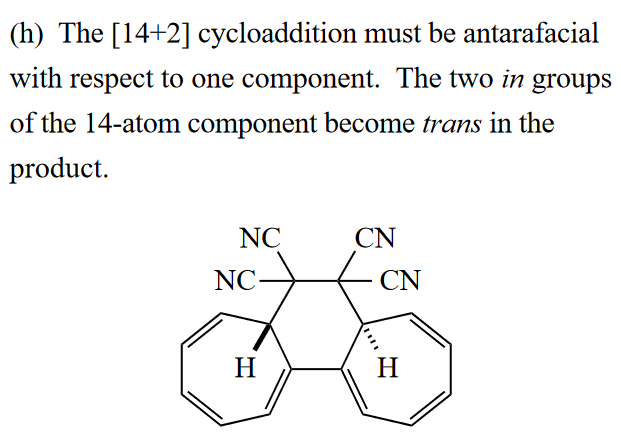
本题为一道[14 + 2]环加成,反应总电子数为16,所以反应是s-a反应,需要经过分子轨道的反转,两个H呈反式。你可能会想,s-a 反应不是位阻很大吗,这能发生吗?事实上,这个反应长期以来被认为是异面环加成的唯一例子,在许多高等有机教材中都有所提及。但是,Izzotti 等人在2022年的**[一篇文章]**指出这个反应也不是协同的,部分摘要如下:
The cycloaddition of heptafulvalene with tetracyanoethylene was previously described as an example of an antarafacial cycloaddition, a [$\pi^{14}{a} + \pi^{2}{s}$] process that afforded only the trans cycloadduct by virtue of the edge-to-face approach of TCNE, facilitated by the S shape of heptafulvalene. The reaction has been investigated in depth and found not to be a concerted antarafacial process.
… …
These studies suggest that edge-to-face interactions are prohibitive in even the most favorable cases.
4. Draw the product of each of the following [3,3] sigmatropic rearrangements, including its stereochemistry. In some cases you may find it necessary to build molecular models in order to see the stereochemical result.
_d06eac7d3ffae.png)
Ans
_23dbf04e5801f.png)
由于一个双键在环上,因此 TS 无法保持常规的椅式过渡态,只能以相对稳定的船式过渡态的形式进行反应。
_209af1b8c2990.png)
Ans
_3fb5036a83e8b.png)
椅式过渡态虽然看似可行,但是会得到一个具有环内反式双键的不稳定产物,因此实际上反应还是通过船式过渡态进行的。
_4abf5322e2481.png)
Ans
_02d4754b4d7c5.png)
由于大环的限制,因此只能选择椅式过渡态。
6. Draw mechanisms for the following reactions.
_17260ddb1226b.png)
Ans
_bcac669273e5b.png)
ene 反应总是容易被忽略,尤其是在复杂反应中。
_f086c0bce29da.png)
Ans
_bf717d66ba3cd.png)
MsCl 在碱作用下生成砜烯(sulfene),其是一个高度活泼的化合物,可以参与DA反应或其他反应。注意其区域选择性,sulfene 的电性可以类比 ketene。
_5eb8c966dc89c.png)
Ans
_0902dee190895.png)
本题的 -NPhth 是一个良好的离去基团,在碱作用下离去后得到 C=S 键,随后发生了 hetero-DA 和 hetero-ene 反应分别得到两个产物。
_c52ec2dc4a17e.png)
Ans
_420f9b8524d5a.png)
反应中第一分子的 LDA 用于拔酰胺 N 上的氢(生成双烯体),第二分子的 LDA 用于生成苯炔(生成亲双烯体),随后发生分子内DA成环。另外答案中也提到了,这个结构相当不稳定,有很强的芳构化倾向。
_20b0de349020b.png)
Ans
_b8b4721534c51.png)
产物中有一分子的丙二酸酯结构,那么不难猜出另一分子的丙二酸酯盐作为碱发挥作用。本题难点在于想到类似 Favorskii 反应的重排。
_b32d32cf8556b.png)
Ans
_fe2fe7d6f88d7.png)
本题第一眼看上去似乎是吲哚双键参与的DA反应,但是发现 C7 是一个季碳中心,无法形成所需的双烯体,因此只能另辟蹊径。加入的催化量 Rh 是为了在 C9 处形成金属卡宾,因而发现正好可以形成 1,3-偶极体,发生偶极环加成后得到产物。
_bb3ac05aa5cba.png)
Ans
_5a17050b77e7e.png)
第二个产物容易想到,其为DA的直接结果。第一个产物就不那么好想得到,仍然是发生了 ene 反应,注意到 ene 反应是把两个 π 键和一个 σ 键转化为两个 σ 键和一个 π 键,因此从反应物的三个双键到产物的一个双键,意味着发生了两遍 ene 反应。
_7da7a1b135806.png)
Ans
_769f3cce09e54.png)
该反应是一个[1,3]-σ重排,根据 Woodward-Hoffmann 规则,热条件的 1,3-σ迁移是异面的,对于 H 原子来说,异面反应在几何上是不可能的。但是对于烷基来说,烷基的 p 轨道可以发生翻转来满足异面反应对称性的要求。本题答案中给的图有点问题,中间体的分子轨道的波相画的不对,正确的图可以参考正文中的图4-49和4-50。
_29dec6f1266ff.png)
Ans
_9634e288c9c59.png)
观察产物结构可以推测出六元环结构应该是通过DA反应构建的,同时产物的 O-S 键的氧原子即为原料的 O12,因此进一步推出可能包含一步重排反应。
_61e20066a6ae8.png)
Ans
_c395dc1ad22e7.png)
题目中的提示说明该产物无法通过苯在光激发下生成,由于周环反应的可逆性,因此说明题示反应路径中也不会有苯出现。光激发条件下,[2+2]环加成是一个可能的选项,反应物在发生[2+2]后,再进行 retro-DA 反应,就能得到6/4/6的三环结构,到了这里,后续的步骤就很显然了,发生一个 6e-电环化关环即可。注意这一系列反应中只有第一步[2+2]需要光条件。
_8f7777a584a36.png)
Ans
_61f18ad4c024b.png)
标号这一步并不难,同时注意到两个叠氮基团都失去了一分子氮气。题目中提示第一步是叠氮的重排反应(类比 Wolff rearrangement or Curtius rearrangement),因此想到第一步酰基重排同时离去一分子氮气。观察产物中 N6-N9 键,因此能够想到第二分子氮气的离去是因为另一个 N 原子的进攻,最后先电环化开环再关环得到产物。
周环反应大概是各类反应中最强调对称性的一类反应了,其反应上的立体选择性也给合成带来了丰富的选择,通过精巧的设计,利用环加成等周环反应可以快速构建出复杂的化合物骨架,这一类看似简单的反应直至今日仍在各种合成工作中发挥关键作用。
第四章结束。